Advanced Tool Development Topics
This tutorial covers some more advanced tool development topics - such as testing and collections. It assumes some basic knowledge about wrapping Galaxy tools and that you have an environment with Planemo available - check out tutorial if you have never developed a Galaxy tool.
Test-Driven Development
An Example Tool - BWA
To get started let’s install BWA. Here we are going to use conda to
install BWA - but however you obtain it should be fine.
$ conda install --force -c conda-forge -c bioconda bwa
... bwa installation ...
$ bwa
Program: bwa (alignment via Burrows-Wheeler transformation)
Version: 0.7.13-r1126
Contact: Heng Li <lh3@sanger.ac.uk>
Usage: bwa <command> [options]
Command: index index sequences in the FASTA format
mem BWA-MEM algorithm
fastmap identify super-maximal exact matches
pemerge merge overlapping paired ends (EXPERIMENTAL)
aln gapped/ungapped alignment
samse generate alignment (single ended)
sampe generate alignment (paired ended)
bwasw BWA-SW for long queries
shm manage indices in shared memory
fa2pac convert FASTA to PAC format
pac2bwt generate BWT from PAC
pac2bwtgen alternative algorithm for generating BWT
bwtupdate update .bwt to the new format
bwt2sa generate SA from BWT and Occ
Note: To use BWA, you need to first index the genome with `bwa index'.
There are three alignment algorithms in BWA: `mem', `bwasw', and
`aln/samse/sampe'. If you are not sure which to use, try `bwa mem'
first. Please `man ./bwa.1' for the manual.
Alternatively you can use Homebrew/linuxbrew to install it:
$ brew install homebrew/science/bwa
Lets start with a simple wrapper for the BWA application (bwa mem in
particular). You can create a new mini-project with a minimal bwa-mem tool
using Planemo’s project_init command.
$ planemo project_init --template bwa bwa
$ cd bwa
This will create a folder with a bwa-mem.xml as follows:
<?xml version="1.0"?>
<tool id="bwa_mem_test" name="Map with BWA-MEM" version="0.0.1">
<description>- map medium and long reads</description>
<requirements>
<requirement type="package" version="0.7.15">bwa</requirement>
<requirement type="package" version="1.3">samtools</requirement>
</requirements>
<command detect_errors="exit_code"><![CDATA[
## Build reference
#set $reference_fasta_filename = "localref.fa"
ln -s "${ref_file}" "${reference_fasta_filename}" &&
bwa index -a is "${reference_fasta_filename}" &&
## Begin BWA-MEM command line
bwa mem
-t "\${GALAXY_SLOTS:-4}"
-v 1 ## Verbosity is set to 1 (errors only)
"${reference_fasta_filename}"
"${fastq_input1}"
| samtools view -Sb - > temporary_bam_file.bam &&
samtools sort -o ${bam_output} temporary_bam_file.bam
]]></command>
<inputs>
<param name="ref_file" type="data" format="fasta" label="Use the following dataset as the reference sequence" help="You can upload a FASTA sequence to the history and use it as reference" />
<param name="fastq_input1" type="data" format="fastqsanger" label="Select fastq dataset" help="Specify dataset with single reads"/>
</inputs>
<outputs>
<data format="bam" name="bam_output" label="${tool.name} on ${on_string} (mapped reads in BAM format)"/>
</outputs>
<tests>
<!-- header describing command-line will always be different -
hence lines_diff="2" on output tag. -->
<test>
<param name="fastq_input1" value="bwa-mem-fastq1.fq" />
<param name="ref_file" value="bwa-mem-mt-genome.fa" />
<output name="bam_output" file="bwa-aln-test1.bam" ftype="bam" lines_diff="2" />
</test>
</tests>
<help>
**BWA MEM options**
Algorithm options::
-k INT minimum seed length [19]
-w INT band width for banded alignment [100]
-d INT off-diagonal X-dropoff [100]
-r FLOAT look for internal seeds inside a seed longer than {-k} * FLOAT [1.5]
-y INT find MEMs longer than {-k} * {-r} with size less than INT [0]
-c INT skip seeds with more than INT occurrences [500]
-D FLOAT drop chains shorter than FLOAT fraction of the longest overlapping chain [0.50]
-W INT discard a chain if seeded bases shorter than INT [0]
-m INT perform at most INT rounds of mate rescues for each read [50]
-S skip mate rescue
-P skip pairing; mate rescue performed unless -S also in use
-e discard full-length exact matches
Scoring options::
-A INT score for a sequence match, which scales options -TdBOELU unless overridden [1]
-B INT penalty for a mismatch [4]
-O INT[,INT] gap open penalties for deletions and insertions [6,6]
-E INT[,INT] gap extension penalty; a gap of size k cost '{-O} + {-E}*k' [1,1]
-L INT[,INT] penalty for 5'- and 3'-end clipping [5,5]
-U INT penalty for an unpaired read pair [17]
Input/output options::
-p first query file consists of interleaved paired-end sequences
-R STR read group header line such as '@RG\tID:foo\tSM:bar' [null]
-v INT verbose level: 1=error, 2=warning, 3=message, 4+=debugging [3]
-T INT minimum score to output [30]
-h INT if there are <INT hits with score >80% of the max score, output all in XA [5]
-a output all alignments for SE or unpaired PE
-C append FASTA/FASTQ comment to SAM output
-V output the reference FASTA header in the XR tag
-Y use soft clipping for supplementary alignments
-M mark shorter split hits as secondary
-I FLOAT[,FLOAT[,INT[,INT]]]
specify the mean, standard deviation (10% of the mean if absent), max
(4 sigma from the mean if absent) and min of the insert size distribution.
FR orientation only. [inferred]
</help>
<citations>
<citation type="doi">10.1093/bioinformatics/btp698</citation>
</citations>
</tool>
Highlighted are two features of Galaxy’s tool XML syntax.
The detect_errors="exit_code" on the command block
will cause Galaxy to use the actual process exit code to determine failure -
in most cases this is superior to the default Galaxy behavior of checking
for the presence of standard error output.
The <citations> block at the bottom will cause Galaxy to generate
exportable citations in the tool form and history UIs.
Improved Input Handling via Test-Driven Development
In this form, the tool only accepts a single input. The first thing we will do is to expand the tool to also allow paired datasets.
Note
Two big ideas behind test-driven development are:
Write a failing test first.
Run the test before you implement the feature. Seeing the initial test failing ensures that your feature is actually being tested.
So let’s start by generating a test output for the two input files (the bootstrapped
example includes two fastq input files to work with bwa-mem-fastq1.fq and
bwa-mem-fastq2.fq). The following commands will create a bwa index on the
fly, map two input files against it, and build and sort a bam output from the
result - all following the pattern from the command block in the tool.
$ cd test-data
$ bwa index -a is bwa-mem-mt-genome.fa
$ bwa mem bwa-mem-mt-genome.fa bwa-mem-fastq1.fq bwa-mem-fastq2.fq | \
samtools view -Sb - > temporary_bam_file.bam && \
(samtools sort -f temporary_bam_file.bam bwa-aln-test2.bam || samtools sort -o bwa-aln-test2.bam temporary_bam_file.bam)
Warning
In many ways this magic is the hardest part of wrapping Galaxy tools and is something this tutorial cannot really teach. The command line magic required for each tool is going to be different. Developing a Galaxy wrapper requires a lot of knowledge of the underlying applications.
Note
Sort appears twice in this odd command because two different versions of samtools with conflicting syntaxes may happen to be on your machine when running this command. Galaxy manages versioned dependencies and so the tool itself does not reflect this complexity.
The primary result of this is the file test-data/bwa-aln-test2.bam. We
will now copy and paste the existing test case to add a new test case that
specifies both fastq inputs as a collection and expects this new output.
<test>
<param name="fastq_input">
<collection type="paired">
<element name="forward" value="bwa-mem-fastq1.fq" />
<element name="reverse" value="bwa-mem-fastq2.fq" />
</collection>
</param>
<param name="ref_file" value="bwa-mem-mt-genome.fa" />
<param name="input_type" value="paired_collection" />
<output name="bam_output" file="bwa-aln-test2.bam" ftype="bam" lines_diff="2" />
</test>
We want to specify the input datasets as a paired collection
(see the collections documentation in this document for more information) and
we need to have a way to allow the user to specify they are
submitting a paired collection instead of a single input. This is where the
fastq_input and input_type variables above came from.
Next run planemo l to verify the tool doesn’t have any
obvious defects. Once the XML is valid - use planemo t
to verify the new test is failing.
$ planemo t
... bunch of output ...
bwa_mem_test[0]: passed
bwa_mem_test[1]: failed
Note
You can run $ firefox tool_test_output.html to see
full details of all executed tests.
Here you can see this second new test is failing - that is good!
The fix is to create a conditional allowing the user to specify an input type.
When modifying the tool and retesting - try passing the --failed flag to
planemo t - it will speed things up by only rerunning tests that have
already failed.
$ planemo t --failed
If you are comfortable with Galaxy tool development - try modifying the tool to make the failing test pass.
- Hint:
You will need to use the
data_collectionparamtype. It accepts many of the same attributes asdataparameters (e.g. seeinput_fastq1) but you will need to specify acollection_typeofpaired.To access the
data_collectionparameter parts in thecommandblock - use$collection_param.forwardand$collection_param.reverse.
Once you get the new test case passing with the --failed parameter - try
running all the tests again to ensure you didn’t break the original test.
$ planemo t
... bunch of output ...
bwa_mem_test[0]: passed
bwa_mem_test[1]: passed
One possible implementation for tests is as follows (sections with changes highlighted).
<?xml version="1.0"?>
<tool id="bwa_mem_test" name="Map with BWA-MEM" version="0.0.1">
<description>- map medium and long reads</description>
<requirements>
<requirement type="package" version="0.7.15">bwa</requirement>
<requirement type="package" version="1.3">samtools</requirement>
</requirements>
<command detect_errors="exit_code"><![CDATA[
## Build reference
#set $reference_fasta_filename = "localref.fa"
ln -s "${ref_file}" "${reference_fasta_filename}" &&
bwa index -a is "${reference_fasta_filename}" &&
## Begin BWA-MEM command line
bwa mem
-t "\${GALAXY_SLOTS:-4}"
-v 1 ## Verbosity is set to 1 (errors only)
"${reference_fasta_filename}"
#set $input_type = $input_type_conditional.input_type
#if $input_type == "single"
"${input_type_conditional.fastq_input1}"
#elif $input_type == "paired_collection"
"${input_type_conditional.fastq_input.forward}" "${input_type_conditional.fastq_input.reverse}"
#end if
| samtools view -Sb - > temporary_bam_file.bam &&
samtools sort -o ${bam_output} temporary_bam_file.bam
]]></command>
<inputs>
<param name="ref_file" type="data" format="fasta" label="Use the following dataset as the reference sequence" help="You can upload a FASTA sequence to the history and use it as reference" />
<conditional name="input_type_conditional">
<param name="input_type" type="select" label="Input Type">
<option value="single" selected="true">Single Dataset</option>
<option value="paired_collection">Paired Collection</option>
</param>
<when value="single">
<param name="fastq_input1" type="data" format="fastqsanger" label="Select fastq dataset" help="Specify dataset with single reads"/>
</when>
<when value="paired_collection">
<param name="fastq_input" format="fastqsanger" type="data_collection" collection_type="paired" label="Select dataset pair" help="Specify paired dataset collection containing paired reads"/>
</when>
</conditional>
</inputs>
<outputs>
<data format="bam" name="bam_output" label="${tool.name} on ${on_string} (mapped reads in BAM format)"/>
</outputs>
<tests>
<!-- header describing command-line will always be different -
hence lines_diff="2" on output tag. -->
<test>
<param name="fastq_input1" value="bwa-mem-fastq1.fq" />
<param name="ref_file" value="bwa-mem-mt-genome.fa" />
<output name="bam_output" file="bwa-aln-test1.bam" ftype="bam" lines_diff="2" />
</test>
<test>
<param name="fastq_input">
<collection type="paired">
<element name="forward" value="bwa-mem-fastq1.fq" />
<element name="reverse" value="bwa-mem-fastq2.fq" />
</collection>
</param>
<param name="ref_file" value="bwa-mem-mt-genome.fa" />
<param name="input_type" value="paired_collection" />
<output name="bam_output" file="bwa-aln-test2.bam" ftype="bam" lines_diff="2" />
</test>
</tests>
<help>
**BWA MEM options**
Algorithm options::
-k INT minimum seed length [19]
-w INT band width for banded alignment [100]
-d INT off-diagonal X-dropoff [100]
-r FLOAT look for internal seeds inside a seed longer than {-k} * FLOAT [1.5]
-y INT find MEMs longer than {-k} * {-r} with size less than INT [0]
-c INT skip seeds with more than INT occurrences [500]
-D FLOAT drop chains shorter than FLOAT fraction of the longest overlapping chain [0.50]
-W INT discard a chain if seeded bases shorter than INT [0]
-m INT perform at most INT rounds of mate rescues for each read [50]
-S skip mate rescue
-P skip pairing; mate rescue performed unless -S also in use
-e discard full-length exact matches
Scoring options::
-A INT score for a sequence match, which scales options -TdBOELU unless overridden [1]
-B INT penalty for a mismatch [4]
-O INT[,INT] gap open penalties for deletions and insertions [6,6]
-E INT[,INT] gap extension penalty; a gap of size k cost '{-O} + {-E}*k' [1,1]
-L INT[,INT] penalty for 5'- and 3'-end clipping [5,5]
-U INT penalty for an unpaired read pair [17]
Input/output options::
-p first query file consists of interleaved paired-end sequences
-R STR read group header line such as '@RG\tID:foo\tSM:bar' [null]
-v INT verbose level: 1=error, 2=warning, 3=message, 4+=debugging [3]
-T INT minimum score to output [30]
-h INT if there are <INT hits with score >80% of the max score, output all in XA [5]
-a output all alignments for SE or unpaired PE
-C append FASTA/FASTQ comment to SAM output
-V output the reference FASTA header in the XR tag
-Y use soft clipping for supplementary alignments
-M mark shorter split hits as secondary
-I FLOAT[,FLOAT[,INT[,INT]]]
specify the mean, standard deviation (10% of the mean if absent), max
(4 sigma from the mean if absent) and min of the insert size distribution.
FR orientation only. [inferred]
</help>
<citations>
<citation type="doi">10.1093/bioinformatics/btp698</citation>
</citations>
</tool>
Note
Exercise: The devteam mappers allow users to specify both a paired
collection or individual datasets (i.e. using two data parameters).
Extend the above conditional to allow that. Remember to write your test
case first and make sure it fails.
Hint: You should not require additional inputs or outputs to do this.
Adding More Parameters
Next up, let’s add some of BWA’s optional parameters to our tool - these
parameters are outlined in the example tool’s help section. To speed this
up and demonstrate another feature of Galaxy - the next test will test the
command-line generated by Galaxy instead of the exact outputs. Not requiring a
complete set of outputs for each test case is convenient because it can speed
development and allows testing more parameter combinations. There are
certain tools and certain parameters where exact outputs are impossible to
pre-determine though.
Lets start with algorithm parameter``-k INT minimum seed length [19]``.
Again, lets do a test first!
<test>
<param name="fastq_input1" value="bwa-mem-fastq1.fq" />
<param name="ref_file" value="bwa-mem-mt-genome.fa" />
<param name="set_algorithm_params" value="true" />
<param name="k" value="20" />
<assert_command>
<has_text text="-k 20" />
</assert_command>
</test>
Continuing our pattern - let’s ensure this new test fails before implementing
the k parameter.
$ planemo t
... bunch of output ...
bwa_mem_test[0]: passed
bwa_mem_test[1]: passed
bwa_mem_test[2]: failed
Reviewing the output - indeed this new test failed as expected (did not
contain expected text ‘-k 20’). Now let’s implement the k parameter and
use planemo t --failed to ensure our implementation is correct.
An example tool with this test and passing.
<?xml version="1.0"?>
<tool id="bwa_mem_test" name="Map with BWA-MEM" version="0.0.1">
<description>- map medium and long reads</description>
<requirements>
<requirement type="package" version="0.7.15">bwa</requirement>
<requirement type="package" version="1.3">samtools</requirement>
</requirements>
<command detect_errors="exit_code"><![CDATA[
## Build reference
#set $reference_fasta_filename = "localref.fa"
ln -s "${ref_file}" "${reference_fasta_filename}" &&
bwa index -a is "${reference_fasta_filename}" &&
## Begin BWA-MEM command line
bwa mem
-t "\${GALAXY_SLOTS:-4}"
-v 1 ## Verbosity is set to 1 (errors only)
#if $algorithm.set_algorithm_params
-k ${algorithm.k}
#end if
"${reference_fasta_filename}"
#set $input_type = $input_type_conditional.input_type
#if $input_type == "single"
"${input_type_conditional.fastq_input1}"
#elif $input_type == "paired_collection"
"${input_type_conditional.fastq_input.forward}" "${input_type_conditional.fastq_input.reverse}"
#end if
| samtools view -Sb - > temporary_bam_file.bam &&
samtools sort -o ${bam_output} temporary_bam_file.bam
]]></command>
<inputs>
<param name="ref_file" type="data" format="fasta" label="Use the following dataset as the reference sequence" help="You can upload a FASTA sequence to the history and use it as reference" />
<conditional name="input_type_conditional">
<param name="input_type" type="select" label="Input Type">
<option value="single" selected="true">Single Dataset</option>
<option value="paired_collection">Paired Collection</option>
</param>
<when value="single">
<param name="fastq_input1" type="data" format="fastqsanger" label="Select fastq dataset" help="Specify dataset with single reads"/>
</when>
<when value="paired_collection">
<param name="fastq_input" format="fastqsanger" type="data_collection" collection_type="paired" label="Select a paired collection" help="Specify paired dataset collection containing paired reads"/>
</when>
</conditional>
<conditional name="algorithm">
<param name="set_algorithm_params" type="boolean" label="Set Algorithm Parameters">
</param>
<when value="true">
<param argument="-k" label="minimum seed length" type="integer" value="19" />
</when>
<when value="false">
</when>
</conditional>
</inputs>
<outputs>
<data format="bam" name="bam_output" label="${tool.name} on ${on_string} (mapped reads in BAM format)"/>
</outputs>
<tests>
<!-- header describing command-line will always be different -
hence lines_diff="2" on output tag. -->
<test>
<param name="fastq_input1" value="bwa-mem-fastq1.fq" />
<param name="ref_file" value="bwa-mem-mt-genome.fa" />
<output name="bam_output" file="bwa-aln-test1.bam" ftype="bam" lines_diff="2" />
</test>
<test>
<param name="fastq_input">
<collection type="paired">
<element name="forward" value="bwa-mem-fastq1.fq" />
<element name="reverse" value="bwa-mem-fastq2.fq" />
</collection>
</param>
<param name="ref_file" value="bwa-mem-mt-genome.fa" />
<param name="input_type" value="paired_collection" />
<output name="bam_output" file="bwa-aln-test2.bam" ftype="bam" lines_diff="2" />
</test>
<test>
<param name="fastq_input1" value="bwa-mem-fastq1.fq" />
<param name="ref_file" value="bwa-mem-mt-genome.fa" />
<param name="set_algorithm_params" value="true" />
<param name="k" value="20" />
<assert_command>
<has_text text="-k 20" />
</assert_command>
</test>
</tests>
<help>
**BWA MEM options**
Algorithm options::
-k INT minimum seed length [19]
-w INT band width for banded alignment [100]
-d INT off-diagonal X-dropoff [100]
-r FLOAT look for internal seeds inside a seed longer than {-k} * FLOAT [1.5]
-y INT find MEMs longer than {-k} * {-r} with size less than INT [0]
-c INT skip seeds with more than INT occurrences [500]
-D FLOAT drop chains shorter than FLOAT fraction of the longest overlapping chain [0.50]
-W INT discard a chain if seeded bases shorter than INT [0]
-m INT perform at most INT rounds of mate rescues for each read [50]
-S skip mate rescue
-P skip pairing; mate rescue performed unless -S also in use
-e discard full-length exact matches
Scoring options::
-A INT score for a sequence match, which scales options -TdBOELU unless overridden [1]
-B INT penalty for a mismatch [4]
-O INT[,INT] gap open penalties for deletions and insertions [6,6]
-E INT[,INT] gap extension penalty; a gap of size k cost '{-O} + {-E}*k' [1,1]
-L INT[,INT] penalty for 5'- and 3'-end clipping [5,5]
-U INT penalty for an unpaired read pair [17]
Input/output options::
-p first query file consists of interleaved paired-end sequences
-R STR read group header line such as '@RG\tID:foo\tSM:bar' [null]
-v INT verbose level: 1=error, 2=warning, 3=message, 4+=debugging [3]
-T INT minimum score to output [30]
-h INT if there are <INT hits with score >80% of the max score, output all in XA [5]
-a output all alignments for SE or unpaired PE
-C append FASTA/FASTQ comment to SAM output
-V output the reference FASTA header in the XR tag
-Y use soft clipping for supplementary alignments
-M mark shorter split hits as secondary
-I FLOAT[,FLOAT[,INT[,INT]]]
specify the mean, standard deviation (10% of the mean if absent), max
(4 sigma from the mean if absent) and min of the insert size distribution.
FR orientation only. [inferred]
</help>
<citations>
<citation type="doi">10.1093/bioinformatics/btp698</citation>
</citations>
</tool>
The tool also demonstrates the new argument option on param tag. These
work a lot like specifying a parameter name argument - but Galaxy will
describe the underlying application argument in the GUI and API - which
may be helpful for power users and external applications.
Exercise 1: Implement a few more algorithm parameters and start
another Scoring section. Extend the above test case as you go.
Exercise 2: Extend the first test case to verify by default none
of these parameters are present in the command. Use the not_has_text
tag to do this (e.g. <not_has_text text="-k 20">).
Exercise 3: Publish the bwa-mem to the local Tool Shed following
the procedure described in the tutorial.
(Don’t forget to alter the commands from the used seqtk example to bwa-mem.)
Hint:
$ planemo shed_init --name=bwa-bwa \
--owner=planemo \
--description=bwa-mem \
--long_description="BWA MEM: Long and medium read mapper" \
--category="Next Gen Mappers"
Note
A full list of the current assertion elements like these that are allowed can be found on the tool syntax page.
In additon to the assertion-based testing of the command, the jobs standard
output and standard error can be checked using assert_stdout and
assert_stderr respectively - paralleling the assert_command tag.
See the sample tool job_properties.xml for an example of this.
Multiple Output Files
Tools which create more than one output file are common. There are several different methods to accommodate this need. Each one of these has their advantages and weaknesses; careful thought should be employed to determine the best method for a particular tool.
Static Multiple Outputs
Handling cases when tools create a static number of outputs is simple. Simply include an <output> tag for each output desired within the tool XML file:
<tool id="example_tool" name="Multiple output" description="example">
<command>example_tool.sh '$input1' $tool_option1 '$output1' '$output2'</command>
<inputs>
...
</inputs>
...
<outputs>
<data format="interval" name="output1" metadata_source="input1" />
<data format="pdf" name="output2" />
</outputs>
</tool>
Static Outputs Determinable from Inputs
In cases when the number of output files varies, but can be determined based upon a user’s parameter selection, the filter tag can be used. The text contents of the <filter> tag are eval``ed and if the expression is ``True a dataset will be created as normal. If the expression is False, the output dataset will not be created; instead a NoneDataset object will be created and made available. When used on the command line the text None will appear instead of a file path. The local namespace of the filter has been populated with the tool parameters.
<tool id="example_tool" name="Multiple output" description="example">
<command>example_tool.sh '$input1' $tool_option1 '$output1' '$output2'</command>
<inputs>
...
<param name="tool_option1" type="select" label="Type of output">
<option value="1">Single File</option>
<option value="2">Two Files</option>
</param>
<conditional name="condition1">
<param name="tool_option2" type="select" label="Conditional output">
<option value="yes">Yes</option>
<option value="no">No</option>
</param>
...
</condition>
...
</inputs>
...
<outputs>
<data format="interval" name="output1" metadata_source="input1" />
<data format="pdf" name="output2" >
<filter>tool_option1 == "2"</filter>
</data>
<data format="txt" name="output3" >
<filter>condition1['tool_option2'] == "yes"</filter>
</data>
</outputs>
</tool>
The command line generated when tool_option1 is set to Single File is:
example_tool.sh input1_FILE_PATH 1 output1_FILE_PATH None
The command line generated when tool_option1 is set to Two Files is:
example_tool.sh input1_FILE_PATH 2 output1_FILE_PATH output2_FILE_PATH
The datatype of an output can be determined by conditional parameter settings as in tools/filter/pasteWrapper.xml
<outputs>
<data format="input" name="out_file1" metadata_source="input1">
<change_format>
<when input_dataset="input1" attribute="ext" value="bed" format="interval"/>
</change_format>
</data>
</outputs>
Single HTML Output
There are times when a single history item is desired, but this history item is composed of multiple files which are only useful when considered together. This is done by having a single (primary) output and storing additional files in a directory (single-level) associated with the primary dataset.
A common usage of this strategy is to have the primary dataset be an HTML file and then store additional content (reports, pdfs, images, etc) in the dataset extra files directory. The content of this directory can be referenced using relative links within the primary (HTML) file, clicking on the eye icon to view the dataset will display the HTML page.
If you want to wrap or create a tool that generates an HTML history item that shows the user links to a number of related output objects (files, images..), you need to know where to write the objects and how to reference them when your tool generates HTML which gets written to the HTML file. Galaxy will not write that HTML for you at present.
The FastQC wrapper is an existing tool example where the Java application generates HTML and image outputs but these need to be massaged to make them Galaxy friendly. In other cases, the application or your wrapper must take care of all the fiddly detailed work of writing valid html to display to the user. In either situation, the html datatype offers a flexible way to display very complex collections of related outputs inside a single history item or to present a complex html page generated by an application. There are some things you need to take care of for this to work:
The following example demonstrates declaring an output of type html.
<outputs>
<data format="html" name="html_file" label="myToolOutput_${tool_name}.html">
</outputs>
The application or script must be set up to write all the output files and/or images to a new special subdirectory passed as a command line parameter from Galaxy every time the tool is run. The paths for images and other files will end up looking something like $GALAXY_ROOT/database/files/000/dataset_56/img1.jpg when you prepend the Galaxy provided path to the filenames you want to use. The command line must pass that path to your script and it is specified using the extra_files_path property of the HTML file output.
For example:
<command>myscript.pl '$input1' '$html_file' '$html_file.extra_files_path' </command>
The application must create and write valid html to setup the page $html_file seen by the user when they view (eye icon) the file. It must create and write that new file at the path passed by Galaxy as the $html_file command line parameter. All application outputs that will be included as links in that html code should be placed in the specific output directory $html_file.extra_files_path passed on the command line. The external application is responsible for creating that directory before writing images and files into it. When generating the html, The files written by the application to $html_file.extra_files_path are referenced in links directly by their name, without any other path decoration - eg:
<a href="file1.xls">Some special output</a>
<br/>
<img src="image1.jpg" >
The (now unmaintained) Galaxy Tool Factory includes code to gather all output files and create a page with links and clickable PDF thumbnail images which may be useful as a starting point (e.g. see rgToolFactory2.py.
galaxy.datatypes.text.Html (the html datatype) is a subclass of composite datasets so new subclasses of composite can be used to implement even more specific structured outputs but this requires adding the new definition to Galaxy - whereas Html files require no extension of the core framework. For more information visit Composite Datatypes.
Dynamic Numbers of Outputs
This section discusses the case where the number of output datasets cannot be determined until the tool run is complete. If the outputs can be broken into groups or collections of similar/homogenous datasets - this is possibly a case for using dataset collections. If instead the outputs should be treated individually and Galaxy’s concept of dataset collections doesn’t map cleanly to the outputs - Galaxy can “discover” individual output datasets dynamically after the job is complete.
Collections
See the Planemo documentation on creating collections for more details on this topic.
A blog post on generating dataset collections from tools can be found here.
Individual Datasets
There are times when the number of output datasets varies entirely based upon the content of an input dataset and the user needs to see all of these outputs as new individual history items rather than as a collection of datasets or a group of related files linked in a single new HTML page in the history. Tools can optionally describe how to “discover” an arbitrary number of files that will be added after the job’s completion to the user’s history as new datasets. Whenever possible, one of the above strategies should be used instead since these discovered datasets cannot be used with workflows and require the user to refresh their history before they are shown.
Discovering datasets (arbitrarily) requires a fixed “parent” output dataset to key on - this dataset will act as the reference for our additional datasets. Sometimes the parent dataset that should be used makes sense from context but in instances where one does not readily make sense tool authors can just create an arbitrary text output (like a report of the dataset generation).
Each discovered dataset requires a unique “designation” (used to describe functional tests, the default output name, etc…) and should be located in the job’s working direcotry or a sub-directory thereof. Regular expressions are used to describe how to discover the datasets and (though not required) a unique such pattern should be specified for each homogeneous group of such files.
Examples
Consider a tool that creates a bunch of text files or bam files and writes them (with extension that matches the Galaxy datatype - e.g. txt or bam to the split sub-directory of the working directory. Such outputs can be discovered by adding the following block of XML to your tool description:
<outputs>
<data name="report" format="txt">
<discover_datasets pattern="__designation_and_ext__" directory="split" visible="true" />
</data>
</outputs>
So for instance, if the tool creates 4 files (in addition to the report) such as split/samp1.bam, split/samp2.bam, split/samp3.bam, and split/samp4.bam - then 4 discovered datasets will be created of type bam with designations of samp1, samp2, samp3, and samp4.
If the tool doesn’t create the files in split with extensions or does but with extensions that do not match Galaxy’s datatypes - a slightly different pattern can be used and the extension/format can be statically specified (here either ext or format may be used as the attribute name):
<outputs>
<data name="report" format="txt">
<discover_datasets pattern="__designation__" format="tabular" directory="tables" visible="true" />
</data>
</outputs>
So in this example, if the tool creates 3 tabular files such as tables/part1.tsv, tables/part2.tsv, and tables/part3.tsv - then 3 discovered datasets will be created of type tabular with designations of part1.tsv, part2.tsv, and part3.tsv.
It may not be desirable for the extension/format (.tsv) to appear in the designation this way. These patterns __designation__ and __designation_and_ext__ are replaced with regular expressions that capture metadata from the file name using named groups. A tool author can explicitly define these regular expressions instead of using these shortcuts - for instance __designation__ is just (?P<designation>.*) and __designation_and_ext__ is (?P<designation>.*)\.(?P<ext>[^\._]+)?. So the above example can be modified as:
<outputs>
<data name="report" format="txt">
<discover_datasets pattern="(?P<designation>.+)\.tsv" format="tabular" directory="tables" visible="true" />
</data>
</outputs>
As a result - three datasets are still be captured - but this time with designations of part1, part2, and part3.
Notice here the < and > in the tool pattern had to be replaced with \< and > to be properly embedded in XML (this is very ugly - apologies).
The metadata elements that can be captured via regular expression named groups this way include ext, designation, name, dbkey, and visible. Each pattern must declare at least either a designation or a name - the other metadata parts ext, dbkey, and visible are all optional and may also be declared explicitly in via attributes on the discover_datasets element (as shown in the above examples).
For tools which do not define a profile version or define one before 16.04, if no discover_datasets element is nested with a tool output - Galaxy will still look for datasets using the named pattern __default__ which expands to primary_DATASET_ID_(?P<designation>[^_]+)_(?P<visible>[^_]+)_(?P<ext>[^_]+)(_(?P<dbkey>[^_]+))?. Many tools use this mechanism as it traditionally was the only way to discover datasets and has the nice advantage of not requiring an explicit declaration and encoding everything (including the output to map to) right in the name of the file itself.
For instance consider the following output declaration:
<outputs>
<data format="interval" name="output1" metadata_source="input1" />
</outputs>
If $output1.id (accessible in the tool command block) is 546 and the tool (likely a wrapper) produces the files primary_546_output2_visible_bed and primary_546_output3_visible_pdf in the job’s working directory - then after execution is complete these two additional datasets (a bed file and a pdf file) are added to the user’s history.
Newer tool profile versions disable this and require the tool author to be more explicit about what files are discovered.
More information
Example tools which demonstrate discovered datasets:
Original pull request for discovered dataset enhancements with implementation details
Legacy information
In the past, it would be necessary to set the attribute force_history_refresh to True to force the user’s history to fully refresh after the tool run has completed. This functionality is now broken and force_history_refresh is ignored by Galaxy. Users now MUST manually refresh their history to see these files. A Trello card used to track the progress on fixing this and eliminating the need to refresh histories in this manner can be found [[https://trello.com/c/f5Ddv4CS/1993-history-api-determine-history-state-running-from-join-on-running-jobs|here]].
Discovered datasets are available via post job hooks (a deprecated feature) by using the designation - e.g. __collected_datasets__['primary'][designation].
In the past these datasets were typically written to $__new_file_path__ instead of the working directory. This is not very scalable and $__new_file_path__ should generally not be used. If you set the option collect_outputs_from in galaxy.ini ensure job_working_directory is listed as an option (if not the only option).
Collections
Galaxy has a concept of dataset collections to group together datasets and operate over them as a single unit.
Galaxy collections are hierarchical and composed from two collection
types - list and paired.
A list is a collection of datasets (or other collections) where each element has an
identifier. Unlike Galaxy dataset names which are transformed throughout complex analyses - theidentifieris generally preserved and can be used for concepts such assamplename that one wants to preserve in the earlier mapping steps of a workflow and use it during reduction steps and reporting later.The paired collection type is much simpler and more specific to sequencing applications. Each
pairedcollection consists of aforwardandreversedataset.
Note
Read more about creating and managing collections.
Composite types include for instance the list:paired collection type -
which represents a list of dataset pairs. In this case, instead of each
dataset having a list idenifier, each pair of datasets does.
Consuming Collections
Many Galaxy tools can be used without modification in conjunction with collections. Galaxy users can take a collection and map over any tool that consumes individual datasets. For instance, early in typical bioinformatics workflows you may have steps that filter raw data, convert to standard formats, perform QC on individual files - users can take lists, pairs, or lists of paired datasets and map over such tools that consume individual dataset (files). Galaxy will then run the tool once for each dataset in the collection and for each output of that tool Galaxy will rebuild a new collection.
Collection elements have the concept an identifier and an index when the collection is created. Both of these are preserved during these mapping steps. As Galaxy builds output collections from these mapping steps, the identifier and index for the output entries match those of the supplied input.

If a tool’s functionality can be applied to individual files in isolation, the implicit mapping described above should be sufficient and no knowledge of collections by tools should be needed. However, tools may need to process multiple files at once - in this case explicit collection consumption is required. This document outlines three cases:
consuming pairs of datasets
consuming lists
consuming arbitrary collections.
Note
If you find yourself consuming a collection of files and calling the underlying application multiple times within the tool command block, you are likely doing something wrong. Just process a pair or a single dataset and allow the user to map over the collection.
Processing Pairs
Dataset collections are not extensively used by typical Galaxy users yet - so
for tools which process paired datasets the recommended best practice is to
allow users to either supply paired collections or two individual datasets.
Furthermore, many tools which process pairs of datasets can also process
single datasets. The following conditional captures this idiom.
<conditional name="fastq_input">
<param name="fastq_input_selector" type="select" label="Single or Paired-end reads" help="Select between paired and single end data">
<option value="paired">Paired</option>
<option value="single">Single</option>
<option value="paired_collection">Paired Collection</option>
<option value="paired_iv">Paired Interleaved</option>
</param>
<when value="paired">
<param name="fastq_input1" type="data" format="fastqsanger" label="Select first set of reads" help="Specify dataset with forward reads"/>
<param name="fastq_input2" type="data" format="fastqsanger" label="Select second set of reads" help="Specify dataset with reverse reads"/>
</when>
<when value="single">
<param name="fastq_input1" type="data" format="fastqsanger" label="Select fastq dataset" help="Specify dataset with single reads"/>
</when>
<when value="paired_collection">
<param name="fastq_input" format="fastqsanger" type="data_collection" collection_type="paired" label="Select a paired collection" label="Select dataset pair" help="Specify paired dataset collection containing paired reads"/>
</when>
</conditional>
This introduces a new param type - data_collection. The optional
attribute collection_type can specify which kinds of
collections are appropriate for this input. Additional data attributes
such as format can further restrict valid collections.
Here we defined that both items of the paired collection must be of datatype
fastqsanger.
In Galaxy’s command block, the individual datasets can be accessed using
$fastq_input1.forward and $fastq_input1.reverse. If processing
arbitrary collection types an array syntax can also be used (e.g.
$fastq_input['forward']).
Note
Mirroring the ability of Galaxy users to map tools that consume individual datasets over lists (and other collection types), users may also map lists of pairs over tools which explicitly consume dataset pair.
If the output of the tool is datasets, the output of this mapping operation (sometimes referred to as subcollection mapping) will be lists. The element identifier and index of the top level of the list will be preserved.

Some example tools which consume paired datasets include:
collection_paired_test (minimal test tool in Galaxy test suite)
Processing Lists (Reductions)
The data_collection parameter type can specify a collection_type or
list but whenever possible, it is recommended to not explicitly
consume lists as a tool author. Parameters of type data can include a
multiple="True" attribute to allow many datasets to be selected
simultaneously. While the default UI will then have Galaxy users pick
individual datasets, they can choose a collections as the tool can
process both. This has the benefit of allowing tools to
process either individual datasets or collections.
A noteworthy difference is that if a parameter of type data with multiple="true" is used, the elements of
the collection are passed to the tool as a (Python) list, i.e. it is not
possible:
to find out if a collection was passed,
to access properties of the collection (name,…), or
to write tests that pass a collection to the parameter (which would allow to name the elements explicitly).
Another drawback is that the ${on_string} of the label contains the list of data sets in the collection (which can be confusing, since these data sets are in most cases hidden) and not the name of the collection.
<param type="data" name="inputs" label="Input BAM(s)" format="bam" multiple="true" />
The command tag can use for loops to build command lines using these parameters.
For instance:
#for $input in $inputs
--input "$input"
#end for
or using the single-line form of this expression:
#for $input in $inputs# $input #end for#
Will produce command strings with an argument for each input (e.g. --input
"/path/to/input1" --input "/path/to/input2"). Other programs may require all
inputs to be supplied in a single parameter. This can be accomplished using
the idiom:
--input "${",".join(map(str, $inputs))}"
Some example tools which consume multiple datasets (including lists) include:
multi_data_param (small test tool in Galaxy test suite)
Also see the tools-devteam repository Pull Request #20 modifying the cufflinks suite of tools for collection compatible reductions.
Processing Identifiers
Collection elements have identifiers that can be used for various kinds of sample
tracking. These identifiers are set when the collection is first created - either
explicitly in the UI (or API), through mapping over collections that preserves input
identifiers, or as the identifier when dynamically discovering collection outputs
described below.
During reduction steps one may likely want to use these - for
reporting, comparisons, etc. When using these multiple data parameters
the dataset objects expose a field called element_identifier. When these
parameters are used with individual datasets - this will just default to being
the dataset’s name, but when used with collections this parameter will be the
element_identifier (i.e. the preserved sample name).
For instance, imagine merging a collection of tabular datasets into a single
table with a new column indicating the sample name the corresponding rows were
derived from using a little fictitious program called merge_rows.
#import re
#for $input in $inputs
merge_rows --name "${re.sub('[^\w\-_]', '_', $input.element_identifier)}" --file "$input" --to $output;
#end for
Note
Here we are rewriting the element identifiers to assure everything is safe to put on the command-line. In the future, collections will not be able to contain keys that are potentially harmful and this won’t be necessary.
Some example tools which utilize element_identifier include:
More on data_collection parameters
The above three cases (users mapping over single tools, consuming pairs, and
consuming lists using multiple data parameters) are hopefully the most
common ways to consume collections for a tool author - but the
data_collection parameter type allows one to handle more cases than just
these.
We have already seen that in command blocks data_collection parameters
can be accessed as arrays by element identifier (e.g.
$input_collection["left"]). This applies for lists and higher-order
structures as well as pairs. The valid element identifiers can be iterated
over using the keys method.
#for $key in $input_collection.keys()
--input_name $key
--input $input_collection[$key]
#end for
#for $input in $input_collection
--input $input
#end for
Importantly, the keys method and direct iteration are both strongly
ordered. If you take a list of files, do a bunch of processing on them to
produce another list, and then consume both collections in a tools - the
elements will match up if iterated over simultaneously.
Finally, if processing arbitrarily nested collections - one can access the
is_collection attribute to determine if a given element is another
collection or just a dataset.
#for $input in $input_collection
--nested ${input.is_collection}
#end for
Some example tools which consume nested collections include:
collection_nested_test (small test tool demonstrating consumption of nested collections)
Creating Collections
Whenever possible simpler operations that produce datasets should be implicitly “mapped over” to produce collections as described above - but there are a variety of situations for which this idiom is insufficient.
Progressively more complex syntax elements exist for the increasingly complex scenarios. Broadly speaking - the three scenarios covered are when the tool produces…
a collection with a static number of elements (mostly for
pairedcollections, but if a tool has fixed binding it might make sense to create a list this way as well)a
listwith the same number of elements as an input list (this would be a common pattern for normalization applications for instance).a
listwhere the number of elements is not knowable until the job is complete.
1. Static Element Count
For this first case - the tool can declare standard data elements below an output collection element in the outputs tag of the tool definition.
<collection name="paired_output" type="paired" label="Split Pair">
<data name="forward" format="txt" />
<data name="reverse" format_source="input1" from_work_dir="reverse.txt" />
</collection>
Templates (e.g. the command tag) can then reference $forward and $reverse or whatever
name the corresponding data elements are given as demonstrated
in test/functional/tools/collection_creates_pair.xml.
The tool should describe the collection type via the type attribute on the collection element.
Data elements can define format, format_source, metadata_source, from_work_dir, and name.
The above syntax would also work for the corner case of static lists. For paired collections specifically however, the type plugin system now knows how to prototype a pair so the following even easier (though less configurable) syntax works.
<collection name="paired_output" type="paired" label="Split Pair" format_source="input1">
</collection>
In this case the command template could then just reference ${paried_output.forward}
and ${paired_output.reverse} as demonstrated in test/functional/tools/collection_creates_pair_from_type.xml.
2. Computable Element Count
For the second case - where the structure of the output is based on the structure of an input - a structured_like attribute can be defined on the collection tag.
<collection name="list_output" type="list" label="Duplicate List" structured_like="input1" inherit_format="true" />
Templates can then loop over input1 or list_output when building up command-line
expressions. See test/functional/tools/collection_creates_list.xml for an example.
format, format_source, and metadata_source can be defined for such collections if the
format and metadata are fixed or based on a single input dataset. If instead the format or metadata
depends on the formats of the collection it is structured like - inherit_format="true" and/or
inherit_metadata="true" should be used instead - which will handle corner cases where there are
for instance subtle format or metadata differences between the elements of the incoming list.
3. Dynamic Element Count
The third and most general case is when the number of elements in a list cannot be determined until runtime. For instance, when splitting up files by various dynamic criteria.
In this case a collection may define one of more discover_dataset elements. As an example of
one such tool that splits a tabular file out into multiple tabular files based on the first
column see test/functional/tools/collection_split_on_column.xml - which includes the following output definition:
<collection name="split_output" type="list" label="Table split on first column">
<discover_datasets pattern="__name_and_ext__" directory="outputs" />
</collection>
Nested Collections
Galaxy Pull Request #538 implemented the ability to define nested output collections. See the pull request and included example tools for more details.
Further Reading
Galaxy Community Conference Talk by John Chilton [Slides][Video].
Pull Request #386 (the initial implementation)
Pull Request #634 (implementing ability for tools to explicitly output collections)
Macros - Reusable Elements
Frequently, tools may require the same XML fragments be repeated in a file (for instance similar conditional branches, repeated options, etc…) or between tools in the same repository (for instance, nearly all of the GATK tools contain the same standard options). Galaxy tools have a macroing system to address this problem.
Direct XML Macros
The following examples are taken from Pull Request 129 the initial implementation of macros. Prior to to the inclusion of macros, the tophat2 wrapper defined several outputs each which had the following identical actions block associated with them:
<actions>
<conditional name="refGenomeSource.genomeSource">
<when value="indexed">
<action type="metadata" name="dbkey">
<option type="from_data_table" name="tophat2_indexes" column="1" offset="0">
<filter type="param_value" column="0" value="#" compare="startswith" keep="False"/>
<filter type="param_value" ref="refGenomeSource.index" column="0"/>
</option>
</action>
</when>
<when value="history">
<action type="metadata" name="dbkey">
<option type="from_param" name="refGenomeSource.ownFile" param_attribute="dbkey" />
</action>
</when>
</conditional>
</actions>
To reuse this action definition, first a macros section has been defined in the tophat2_wrpper.xml file.
<tool>
...
<macros>
<xml name="dbKeyActions">
<action><!-- Whole big example above. -->
....
</action>
</xml>
</macros>
With this in place, each output data element can include this block using the expand XML element as follows:
<outputs>
<data format="bed" name="insertions" label="${tool.name} on ${on_string}: insertions" from_work_dir="tophat_out/insertions.bed">
<expand macro="dbKeyActions" />
</data>
<data format="bed" name="deletions" label="${tool.name} on ${on_string}: deletions" from_work_dir="tophat_out/deletions.bed">
<expand macro="dbKeyActions" />
</data>
<data format="bed" name="junctions" label="${tool.name} on ${on_string}: splice junctions" from_work_dir="tophat_out/junctions.bed">
<expand macro="dbKeyActions" />
</data>
<data format="bam" name="accepted_hits" label="${tool.name} on ${on_string}: accepted_hits" from_work_dir="tophat_out/accepted_hits.bam">
<expand macro="dbKeyActions" />
</data>
</outputs>
This has reduced the size of the XML file by dozens of lines and reduces the long term maintenance associated with copied and pasted code.
Imported Macros
The macros element described above, can also contain any number of
import elements. This allows a directory/repository of tool XML files to
contain shared macro definitions that can be used by any number of actual tool
files in that directory/repository.
Revisiting the tophat example, all three tophat wrappers (tophat_wrapper.xml,
tophat_color_wrapper.xml, and tophat2_wrapper.xml) shared some common
functionality. To reuse XML elements between these files, a
tophat_macros.xml file was added to that directory.
The following block is a simplified version of that macros file’s contents:
<macros>
<xml name="own_junctionsConditional">
<conditional name="own_junctions">
<param name="use_junctions" type="select" label="Use Own Junctions">
<option value="No">No</option>
<option value="Yes">Yes</option>
</param>
<when value="Yes">
<conditional name="gene_model_ann">
<param name="use_annotations" type="select" label="Use Gene Annotation Model">
<option value="No">No</option>
<option value="Yes">Yes</option>
</param>
<when value="No" />
<when value="Yes">
<param format="gtf,gff3" name="gene_annotation_model" type="data" label="Gene Model Annotations" help="TopHat will use the exon records in this file to build a set of known splice junctions for each gene, and will attempt to align reads to these junctions even if they would not normally be covered by the initial mapping."/>
</when>
</conditional>
<expand macro="raw_juncsConditional" />
<expand macro="no_novel_juncsParam" />
</when>
<when value="No" />
</conditional> <!-- /own_junctions -->
</xml>
<xml name="raw_juncsConditional">
<conditional name="raw_juncs">
<param name="use_juncs" type="select" label="Use Raw Junctions">
<option value="No">No</option>
<option value="Yes">Yes</option>
</param>
<when value="No" />
<when value="Yes">
<param format="interval" name="raw_juncs" type="data" label="Raw Junctions" help="Supply TopHat with a list of raw junctions. Junctions are specified one per line, in a tab-delimited format. Records look like: [chrom] [left] [right] [+/-] left and right are zero-based coordinates, and specify the last character of the left sequenced to be spliced to the first character of the right sequence, inclusive."/>
</when>
</conditional>
</xml>
<xml name="no_novel_juncsParam">
<param name="no_novel_juncs" type="select" label="Only look for supplied junctions">
<option value="No">No</option>
<option value="Yes">Yes</option>
</param>
</xml>
</macros>
Any tool definition in that directory can use the macros contained therein once imported as shown below.
<tool>
...
<macros>
<import>tophat_macros.xml</import>
</macros>
...
<inputs>
<expand macro="own_junctionsConditional" />
...
</inputs>
...
</tool>
This example also demonstrates that macros may themselves expand macros.
Parameterizing XML Macros (with yield)
In some cases, tools may contain similar though not exact same definitions. Some parameterization can be performed by declaring expand elements with child elements and expanding them in the macro definition with a yield element.
For instance, previously the tophat wrapper contained the following definition:
<conditional name="refGenomeSource">
<param name="genomeSource" type="select" label="Will you select a reference genome from your history or use a built-in index?" help="Built-ins were indexed using default options">
<option value="indexed">Use a built-in index</option>
<option value="history">Use one from the history</option>
</param>
<when value="indexed">
<param name="index" type="select" label="Select a reference genome" help="If your genome of interest is not listed, contact the Galaxy team">
<options from_data_table="tophat_indexes_color">
<filter type="sort_by" column="2"/>
<validator type="no_options" message="No indexes are available for the selected input dataset"/>
</options>
</param>
</when>
<when value="history">
<param name="ownFile" type="data" format="fasta" metadata_name="dbkey" label="Select the reference genome" />
</when> <!-- history -->
</conditional> <!-- refGenomeSource -->
and the tophat2 wrapper contained the highly analogous definition:
<conditional name="refGenomeSource">
<param name="genomeSource" type="select" label="Will you select a reference genome from your history or use a built-in index?" help="Built-ins were indexed using default options">
<option value="indexed">Use a built-in index</option>
<option value="history">Use one from the history</option>
</param>
<when value="indexed">
<param name="index" type="select" label="Select a reference genome" help="If your genome of interest is not listed, contact the Galaxy team">
<options from_data_table="tophat2_indexes_color">
<filter type="sort_by" column="2"/>
<validator type="no_options" message="No indexes are available for the selected input dataset"/>
</options>
</param>
</when>
<when value="history">
<param name="ownFile" type="data" format="fasta" metadata_name="dbkey" label="Select the reference genome" />
</when> <!-- history -->
</conditional> <!-- refGenomeSource -->
These blocks differ only in the from_data_table attribute on the options element. To capture this pattern, tophat_macros.xml contains the following macro definition:
<xml name="refGenomeSourceConditional">
<conditional name="refGenomeSource">
<param name="genomeSource" type="select" label="Use a built in reference genome or own from your history" help="Built-ins genomes were created using default options">
<option value="indexed" selected="True">Use a built-in genome</option>
<option value="history">Use a genome from history</option>
</param>
<when value="indexed">
<param name="index" type="select" label="Select a reference genome" help="If your genome of interest is not listed, contact the Galaxy team">
<yield />
</param>
</when>
<when value="history">
<param name="ownFile" type="data" format="fasta" metadata_name="dbkey" label="Select the reference genome" />
</when> <!-- history -->
</conditional> <!-- refGenomeSource -->
</xml>
Notice the yield statement in lieu of an options declaration. This allows the nested options element to be declared when expanding the macro:
The following expand declarations have replaced the original conditional elements.
<expand macro="refGenomeSourceConditional">
<options from_data_table="tophat_indexes">
<filter type="sort_by" column="2"/>
<validator type="no_options" message="No genomes are available for the selected input dataset"/>
</options>
</expand>
<expand macro="refGenomeSourceConditional">
<options from_data_table="tophat2_indexes">
<filter type="sort_by" column="2"/>
<validator type="no_options" message="No genomes are available for the selected input dataset"/>
</options>
</expand>
From Galaxy release 22.01 named yields are supported. That is, if the macro contains
<yield name="xyz"/> it is replaced by the content of the token element with the same name.
Token elements need to be direct children of the expand element. This is useful if different
parts of the macro should be parametrized.
In the following example two named yield and one unnamed yield are used to parametrize the options of the select of a conditional, the options of another select, and additional when block(s). Given the following macro:
<xml name="named_yields_example">
<conditional>
<param type="select">
<option value="a">A</option>
<option value="b">B</option>
<yield name="more_options"/>
</param>
<when value="a">
<param name="aselect" type="select">
<yield />
</param>
</when>
<when value="b"/>
<yield name="more_whens">
</conditional>
</xml>
and expanding the macro in the following way:
<expand macro="named_yields_example">
<token name="more_options">
<option value="c">C</option>
</token>
<token name="more_whens">
<when value="c">
<param type="select">
<yield />
</param>
</when>
</token>
<options from_data_table="tophat2_indexes">
<filter type="sort_by" column="2"/>
<validator type="no_options" message="No genomes are available for the selected input dataset"/>
</options>
</expand>
we get the following expanded definition:
<xml name="named_yields_example">
<conditional>
<param type="select">
<option value="a">A</option>
<option value="b">B</option>
<option value="c">C</option>
</param>
<when value="a">
<param name="aselect" type="select">
<options from_data_table="tophat2_indexes">
<filter type="sort_by" column="2"/>
<validator type="no_options" message="No genomes are available for the selected input dataset"/>
</options>
</param>
</when>
<when value="b"/>
<when value="c">
<param type="select">
<options from_data_table="tophat2_indexes">
<filter type="sort_by" column="2"/>
<validator type="no_options" message="No genomes are available for the selected input dataset"/>
</options>
</param>
</when>
</conditional>
</xml>
Named yields are replaced in the order of the tokens defined in the expand
tag. Unnamed yields are replaced after all named tokens have been replaced (by
the non-token child elements of the expand tag). If there are named
yields that have no corresponding token, then they are treated like unnamed
yields. Note that unnamed and named tokens can be used multiple times in a
macro, then each occurrence is replaced by the corresponding content defined in
the expand.
Further, note that the order of the replacements offers some possibilities
to achieve recursion-like replacements, since a token may contain further named
or unnamed yield tags (see for instance the yield tag contained in the
named token more_whens).
Parameterizing XML Macros (with tokens)
In addition to using yield blocks, there is another way to parametrize
macros by specifying:
required parameters as comma-separated list of parameter names using the
tokensattribute (e.g.tokens="foo,bar") of thexmlelement and then using@FOO@and@BAR@in the macro definition;optional parameters as
token_xyz="default_value"attributes of thexmlelement, and then using@XYZ@in the macro definition.
<macros>
<xml name="color" tokens="varname" token_default_color="#00ff00" token_label="Pick a color">
<param name="@VARNAME@" type="color" label="@LABEL@" value="@DEFAULT_COLOR@" />
</xml>
</macros>
This defines a macro with a required parameter varname and two optional
parameters default_color and label. When invoking this macro, you can
pass values for those parameters and produce varying results.
<inputs>
<expand macro="color" varname="myvar" default_color="#ff0000" />
<expand macro="color" varname="c2" default_color="#0000ff" label="Choose a different color" />
</inputs>
The attributes passed to the macro definition will be filled in (or defaults used if not provided). Effectively this yields:
<inputs>
<param name="myvar" type="color" label="Pick a color" value="#ff0000" />
<param name="c2" type="color" label="Choose a different color" value="#0000ff" />
</inputs>
Macro tokens can be used in the text content of tags, attribute values, and (with a
little trick also in attribute names). The problem is that the default delimiting character of
macro tokens is @ and the XML must still be valid before processing the macros (and @
is invalid in attribute names). Luckily the delimiting character(s) can be changed by adding
token_quote to the macro definition:
<macros>
<xml name="color" tokens="attr,attr_value" token_quote="__" token_label="label">
<param __ATTR__="__ATTR_VALUE__" label="__LABEL__"/>
</xml>
</macros>
Note that, this forbids to use tokens with the name quote.
Macro Tokens
You can use
<token name="@IS_PART_OF_VCFLIB@">is a part of VCFlib toolkit developed by Erik Garrison (https://github.com/ekg/vcflib).</token>
and then call the token within any element of the file like this:
Vcfallelicprimitives @IS_PART_OF_VCFLIB@
Tool Provided Metadata
This stub of a section provides some initial documentation on tool provided metadata. Galaxy allows datasets to be discovered after a tool has been executed and allows tools to specify metadata for these datasets. Whenever possible, Galaxy’s datatypes and more structured outputs should be used to collect metadata.
If an arbitrary number of outputs is needed but no special metadata must be set, file name patterns can be used to allow Galaxy to discover these datasets. More information on this can be found in the dedicated section.
The file name patterns described in the above link are nice because they don’t need special instrumenting in the tool wrapper to adapt to Galaxy in general and can adapt to many existing application’s output. When more metadata must be supplied or when implementing a custom tool wrapper anyway - it may be beneficial to build a manifest file.
A tool may also produce a file called galaxy.json during execution. If
upon a job’s completion this file is populated, Galaxy will expect to find metadata
about outputs in it.
The format of this file is a bit quirky - each line of this file should be a JSON
dictionary. Each such dictionary should contain a type attribute - this type
may be either new_primary_dataset or dataset.
If the type is new_primary_dataset, the dictionary should contain a
filename entry with a path to a “discovered dataset”. In this case the
dictionary may contain any of the following entries name, dbkey, info, ext, metadata.
namewill be used as the output dataset’s nameextallows specification of the format of the output (e.g.txt,tabular,fastqsanger, etc…)dbkeyallows specifying a genome build for the discovered datasetinfois a short text description for each dataset that appears in the history panelmetadatathis should be a dictionary of key-value pairs for metadata registered with the datatype for this output
Examples of tools using new_primary_dataset entries:
tool_provided_metadata_2.xml demonstrating using the simpler attributes described here.
tool_provided_metadata_3.xml demonstrates overridding datatype specified metadata.
The type of an entry may also be dataset. In this case the metadata
descriptions describe an explicit output (one with its own corresponding output
element definition). In this case, an entry called dataset should appear in
the dictionary (in lieu of filename above) and should be the database id of the
output dataset. Such entries may contain all of the other fields described above except
metadata.
Example tool using a dataset entry:
Cluster Usage
Developing for Clusters - GALAXY_SLOTS, GALAXY_MEMORY_MB, and GALAXY_MEMORY_MB_PER_SLOT
GALAXY_SLOTS is a special environment variable that is set in a Galaxy
tool’s runtime environment. If the tool you are working on allows configuring
the number of processes or threads that should be spawned, this variable
should be used.
For example, for the StringTie tool
the binary stringtie can take an argument -p that allows specification
of the number of threads to be used. The Galaxy tool sets this up as follows
stringtie "$input_bam" -o "$output_gtf" -p "\${GALAXY_SLOTS:-1}" ...
Here we use \${GALAXY_SLOTS:-Z} instead of a fixed value (Z being an
integer representing a default value in non-Galaxy contexts). The
backslash here is because this value is interpreted at runtime as
environment variable - not during command building time as a templated
value. Now server administrators can configure how many processes the
tool should be allowed to use.
For information on how server administrators can configure this value for a particular tool, check out the Galaxy admin documentation.
Analogously GALAXY_MEMORY_MB and GALAXY_MEMORY_MB_PER_SLOT are special
environment variables in a Galaxy tool’s runtime environment that can be used
to specify the amount of memory that a tool can use overall and per slot,
respectively.
For an example see the samtools sort tool which allows to specify the total memory with the -m parameter.
Test Against Clusters - --job_config_file
The various commands that start Galaxy servers (serve,
test, shed_serve, shed_test, etc…) allow specification of
a Galaxy job configuration XML file (e.g. job_conf.xml).
For instance, Slurm is a popular distributed reource manager (DRM) in the
Galaxy community. The following job_conf.xml tells Galaxy to run all jobs
using Slurm and allocate 2 cores for each job.
<?xml version="1.0"?>
<job_conf>
<plugins>
<plugin id="drmaa" type="runner" load="galaxy.jobs.runners.drmaa:DRMAAJobRunner" />
</plugins>
<handlers>
<handler id="main"/>
</handlers>
<destinations default="drmaa">
<destination id="drmaa" runner="drmaa">
<param id="nativeSpecification">--time=00:05:00 --nodes=1 --ntasks=2</param>
</destination>
</destinations>
</job_conf>
If this file is named planemo_job_conf.xml and resides in one’s home
directory (~), Planemo can test or serve using this configuration
with the following commands.
$ planemo test --job_config_file ~/planemo_job_conf.xml .
$ planemo serve --job_config_file ~/planemo_job_conf.xml .
For general information on configuring Galaxy to communicate with clusters check out this page on the Galaxy wiki and for information regarding configuring job configuration XML files in particular check out the example distributed with Galaxy.
Dependencies and Conda
Specifying and Using Tool Requirements
Note
This document discusses using Conda to satisfy tool dependencies from a tool developer perspective. An in depth discussion of using Conda to satisfy dependencies from an admistrator’s perspective can be found here. That document also serves as good background for this discussion.
Note
Planemo requires a Conda installation to target with its various Conda
related commands. A properly configured Conda installation can be initialized
with the conda_init command. This should only need to be executed once
per development machine.
$ planemo conda_init
galaxy.tools.deps.conda_util INFO: Installing conda, this may take several minutes.
wget -q --recursive -O /var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/conda_installLW5zn1.sh https://repo.continuum.io/miniconda/Miniconda3-4.3.31-MacOSX-x86_64.sh
bash /var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/conda_installLW5zn1.sh -b -p /Users/john/miniconda3
PREFIX=/Users/john/miniconda3
installing: python-3.6.3-h47c878a_7 ...
Python 3.6.3 :: Anaconda, Inc.
installing: ca-certificates-2017.08.26-ha1e5d58_0 ...
installing: conda-env-2.6.0-h36134e3_0 ...
installing: libcxxabi-4.0.1-hebd6815_0 ...
installing: tk-8.6.7-h35a86e2_3 ...
installing: xz-5.2.3-h0278029_2 ...
installing: yaml-0.1.7-hc338f04_2 ...
installing: zlib-1.2.11-hf3cbc9b_2 ...
installing: libcxx-4.0.1-h579ed51_0 ...
installing: openssl-1.0.2n-hdbc3d79_0 ...
installing: libffi-3.2.1-h475c297_4 ...
installing: ncurses-6.0-hd04f020_2 ...
installing: libedit-3.1-hb4e282d_0 ...
installing: readline-7.0-hc1231fa_4 ...
installing: sqlite-3.20.1-h7e4c145_2 ...
installing: asn1crypto-0.23.0-py36h782d450_0 ...
installing: certifi-2017.11.5-py36ha569be9_0 ...
installing: chardet-3.0.4-py36h96c241c_1 ...
installing: idna-2.6-py36h8628d0a_1 ...
installing: pycosat-0.6.3-py36hee92d8f_0 ...
installing: pycparser-2.18-py36h724b2fc_1 ...
installing: pysocks-1.6.7-py36hfa33cec_1 ...
installing: python.app-2-py36h54569d5_7 ...
installing: ruamel_yaml-0.11.14-py36h9d7ade0_2 ...
installing: six-1.11.0-py36h0e22d5e_1 ...
installing: cffi-1.11.2-py36hd3e6348_0 ...
installing: setuptools-36.5.0-py36h2134326_0 ...
installing: cryptography-2.1.4-py36h842514c_0 ...
installing: wheel-0.30.0-py36h5eb2c71_1 ...
installing: pip-9.0.1-py36h1555ced_4 ...
installing: pyopenssl-17.5.0-py36h51e4350_0 ...
installing: urllib3-1.22-py36h68b9469_0 ...
installing: requests-2.18.4-py36h4516966_1 ...
installing: conda-4.3.31-py36_0 ...
installation finished.
/Users/john/miniconda3/bin/conda install -y --override-channels --channel iuc --channel conda-forge --channel bioconda --channel defaults conda=4.3.33 conda-build=2.1.18
Fetching package metadata ...................
Solving package specifications: .
Package plan for installation in environment /Users/john/miniconda3:
The following NEW packages will be INSTALLED:
beautifulsoup4: 4.6.0-py36_0 conda-forge
conda-build: 2.1.18-py36_0 conda-forge
conda-verify: 2.0.0-py36_0 conda-forge
filelock: 3.0.4-py36_0 conda-forge
jinja2: 2.10-py36_0 conda-forge
markupsafe: 1.0-py36_0 conda-forge
pkginfo: 1.4.2-py36_0 conda-forge
pycrypto: 2.6.1-py36_1 conda-forge
pyyaml: 3.12-py36_1 conda-forge
The following packages will be UPDATED:
conda: 4.3.31-py36_0 --> 4.3.33-py36_0 conda-forge
beautifulsoup4 100% |###################################################################| Time: 0:00:00 782.08 kB/s
filelock-3.0.4 100% |###################################################################| Time: 0:00:00 7.95 MB/s
markupsafe-1.0 100% |###################################################################| Time: 0:00:00 5.82 MB/s
pkginfo-1.4.2- 100% |###################################################################| Time: 0:00:00 1.18 MB/s
pycrypto-2.6.1 100% |###################################################################| Time: 0:00:00 1.69 MB/s
pyyaml-3.12-py 100% |###################################################################| Time: 0:00:00 3.31 MB/s
conda-verify-2 100% |###################################################################| Time: 0:00:00 6.91 MB/s
jinja2-2.10-py 100% |###################################################################| Time: 0:00:00 2.81 MB/s
conda-4.3.33-p 100% |###################################################################| Time: 0:00:00 621.27 kB/s
conda-build-2. 100% |###################################################################| Time: 0:00:00 2.16 MB/s
Conda installation succeeded - Conda is available at '/Users/john/miniconda3/bin/conda'
While Galaxy can be configured to resolve dependencies various ways, Planemo is configured with opinionated defaults geared at making building tools that target Conda as easy as possible.
During the introductory tool development tutorial, we called planemo tool_init
with the argument --requirement seqtk@1.2 and the resulting tool contained
the XML:
<requirements>
<requirement type="package" version="1.2">seqtk</requirement>
</requirements>
As configured by Planemo, when Galaxy encounters these requirement tags it
will attempt to install Conda, check for referenced packages (such as
seqtk), and install them as needed for tool testing.
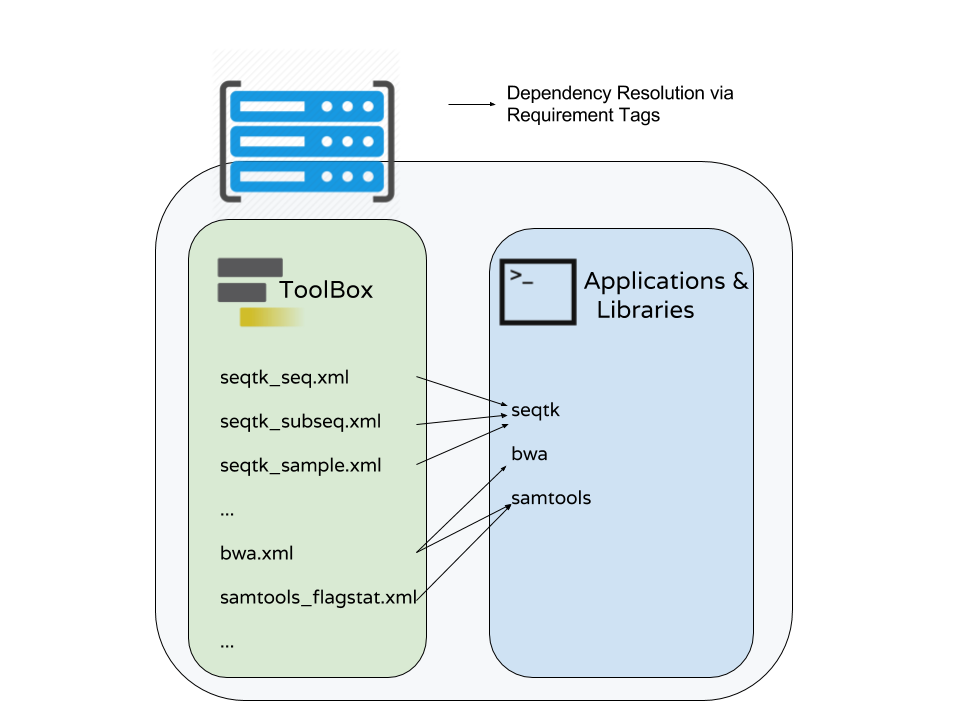
Galaxy’s dependency resolution maps tool requirement tags to concrete applications and libraries setup by the Galaxy deployer (or Planemo). As the above diagram indicates the same requirements may be used by multiple Galaxy tools and a single Galaxy tool may depend on multiple requirements. The document describes working with Conda dependencies from a developer perspective but other dependency resolution techniques are covered in the Galaxy docs.
Note
Why Conda?
Many different package managers could potentially be targeted here, but we focus on Conda for a few key reasons.
No compilation at install time - binaries with their dependencies and libraries
Support for all operating systems
Easy to manage multiple versions of the same recipe
HPC-ready: no root privileges needed
Easy-to-write YAML recipes
Viberant communities
Note
Conda Terminology
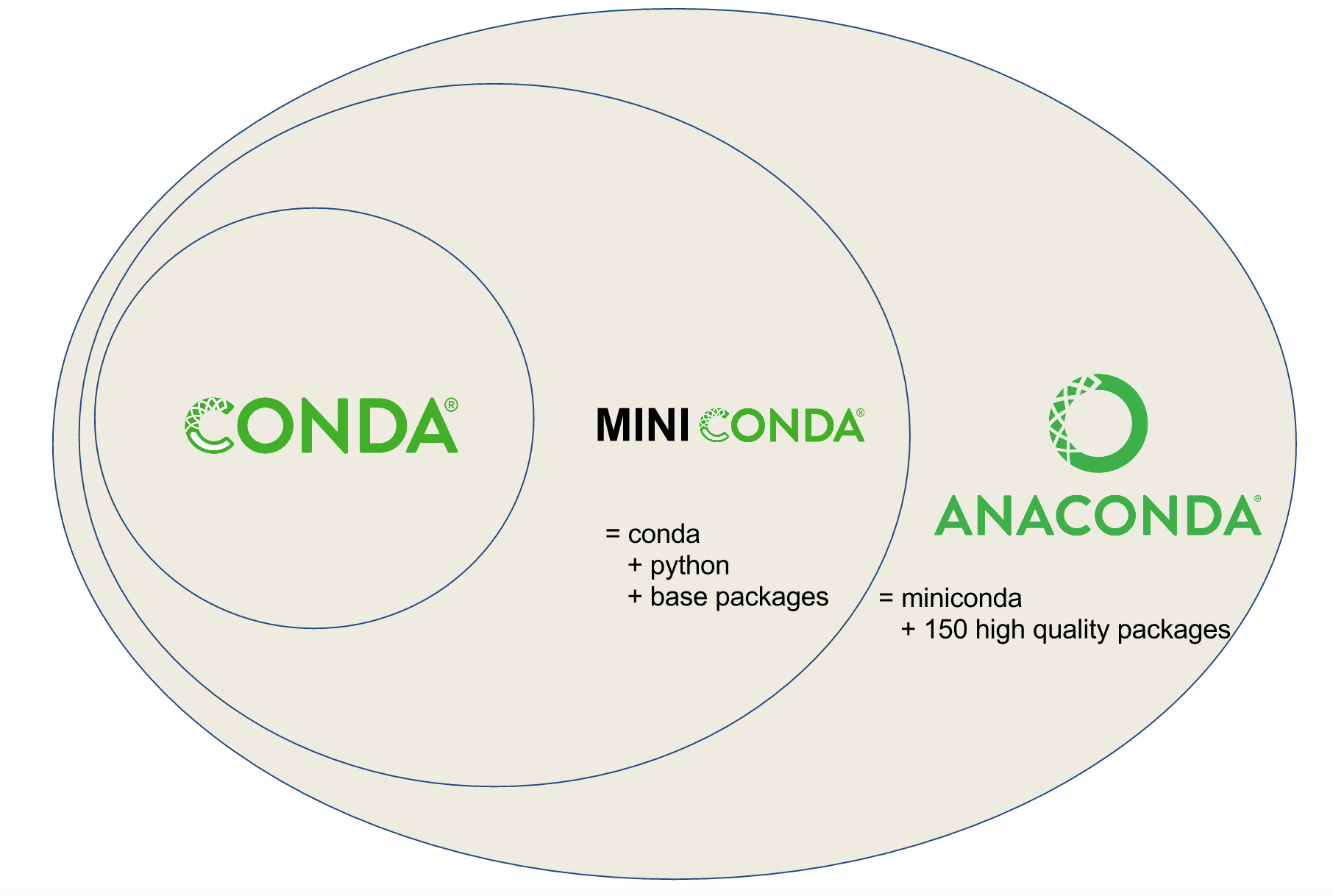
Conda recipes build packages that are published to channels.
Planemo is setup to target a few channels by default, these include iuc, bioconda,
conda_forge, defaults - the whole dependency management scheme outlined here works a lot
better if packages can be found in one of these “best practice” channels.
We can check if the requirements on a tool are available in best practice
Conda channels using an extended form of the planemo lint command. Passing
--conda_requirements flag will ensure all listed requirements are found.
$ planemo lint --conda_requirements seqtk_seq.xml
Linting tool /Users/john/workspace/planemo/docs/writing/seqtk_seq.xml
...
Applying linter requirements_in_conda... CHECK
.. INFO: Requirement [seqtk@1.2] matches target in best practice Conda channel [bioconda].
Note
You can download the final version of the seqtk seq wrapper from the Planemo tutorial using the command:
$ planemo project_init --template=seqtk_complete seqtk_example
$ cd seqtk_example
We can verify these tool requirements install with the conda_install command. With
its default parameters conda_install processes tools and creates isolated environments
for their declared requirements.
$ planemo conda_install seqtk_seq.xml
Install conda target CondaTarget[seqtk,version=1.2]
/home/john/miniconda2/bin/conda create -y --name __seqtk@1.2 seqtk=1.2
Fetching package metadata ...............
Solving package specifications: ..........
Package plan for installation in environment /home/john/miniconda2/envs/__seqtk@1.2:
The following packages will be downloaded:
package | build
---------------------------|-----------------
seqtk-1.2 | 0 29 KB bioconda
The following NEW packages will be INSTALLED:
seqtk: 1.2-0 bioconda
zlib: 1.2.8-3
Fetching packages ...
seqtk-1.2-0.ta 100% |#############################################################| Time: 0:00:00 444.71 kB/s
Extracting packages ...
[ COMPLETE ]|################################################################################| 100%
Linking packages ...
[ COMPLETE ]|################################################################################| 100%
#
# To activate this environment, use:
# > source activate __seqtk@1.2
#
# To deactivate this environment, use:
# > source deactivate __seqtk@1.2
#
$ which seqtk
seqtk not found
$
The above install worked properly, but seqtk is not on your PATH because this merely
created an environment within the Conda directory for the seqtk installation. Planemo
will configure Galaxy to exploit this installation. If you wish to interactively explore
the resulting enviornment to explore the installed tool or produce test data the output
of the conda_env command can be sourced.
$ . <(planemo conda_env seqtk_seq.xml)
Deactivate environment with conda_env_deactivate
(seqtk_seq) $ which seqtk
/home/planemo/miniconda2/envs/jobdepsiJClEUfecc6d406196737781ff4456ec60975c137e04884e4f4b05dc68192f7cec4656/bin/seqtk
(seqtk_seq) $ seqtk seq
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
-U convert all bases to uppercases
-S strip of white spaces in sequences
(seqtk_seq) $ conda_env_deactivate
$
As shown above the conda_env_deactivate will be created in this environment and can
be used to restore your initial shell configuration.
Confident the underlying application works, we can now use planemo test or
planemo serve and it will reuse this environment and find our dependency (in this
case seqtk as needed).
Here is a portion of the output from the testing command planemo test seqtk_seq.xml
demonstrating using this tool.
$ planemo test seqtk_seq.xml
...
2017-02-22 10:13:28,902 INFO [galaxy.tools.actions] Handled output named output1 for tool seqtk_seq (20.136 ms)
2017-02-22 10:13:28,914 INFO [galaxy.tools.actions] Added output datasets to history (12.782 ms)
2017-02-22 10:13:28,935 INFO [galaxy.tools.actions] Verified access to datasets for Job[unflushed,tool_id=seqtk_seq] (10.954 ms)
2017-02-22 10:13:28,936 INFO [galaxy.tools.actions] Setup for job Job[unflushed,tool_id=seqtk_seq] complete, ready to flush (21.053 ms)
2017-02-22 10:13:28,962 INFO [galaxy.tools.actions] Flushed transaction for job Job[id=2,tool_id=seqtk_seq] (26.510 ms)
2017-02-22 10:13:29,064 INFO [galaxy.jobs.handler] (2) Job dispatched
2017-02-22 10:13:29,281 DEBUG [galaxy.tools.deps] Using dependency seqtk version 1.2 of type conda
2017-02-22 10:13:29,282 DEBUG [galaxy.tools.deps] Using dependency seqtk version 1.2 of type conda
2017-02-22 10:13:29,317 INFO [galaxy.jobs.command_factory] Built script [/tmp/tmpLvKwta/job_working_directory/000/2/tool_script.sh] for tool command [[ "$CONDA_DEFAULT_ENV" = "/Users/john/miniconda2/envs/__seqtk@1.2" ] || . /Users/john/miniconda2/bin/activate '/Users/john/miniconda2/envs/__seqtk@1.2' >conda_activate.log 2>&1 ; seqtk seq -A '/tmp/tmpLvKwta/files/000/dataset_1.dat' > '/tmp/tmpLvKwta/files/000/dataset_2.dat']
2017-02-22 10:13:29,516 DEBUG [galaxy.tools.deps] Using dependency samtools version None of type conda
2017-02-22 10:13:29,516 DEBUG [galaxy.tools.deps] Using dependency samtools version None of type conda
ok
----------------------------------------------------------------------
XML: /private/tmp/tmpLvKwta/xunit.xml
----------------------------------------------------------------------
Ran 1 test in 15.936s
OK
2017-02-22 10:13:37,014 INFO [test_driver] Shutting down
2017-02-22 10:13:37,014 INFO [test_driver] Shutting down embedded galaxy web server
2017-02-22 10:13:37,016 INFO [test_driver] Embedded web server galaxy stopped
2017-02-22 10:13:37,017 INFO [test_driver] Stopping application galaxy
....
2017-02-22 10:13:37,018 INFO [galaxy.jobs.handler] sending stop signal to worker thread
2017-02-22 10:13:37,018 INFO [galaxy.jobs.handler] job handler stop queue stopped
Testing complete. HTML report is in "/Users/john/workspace/planemo/project_templates/seqtk_complete/tool_test_output.html".
All 1 test(s) executed passed.
seqtk_seq[0]: passed
In this case the tests passed and the line containing [galaxy.tools.deps] Using dependency seqtk version 1.2 of type conda
indicates Galaxy dependency resolution was successful and it found the environment we previously installed with conda_install.
Finding Existing Conda Packages
How did we know what software name and software version to use? We found the existing
packages available for Conda and referenced them. To do this yourself, you can simply
use the planemo command conda_search. If we do a search for seqt it will show
all the software and all the versions available matching that search term - including
seqtk.
$ planemo conda_search seqt
/Users/john/miniconda3/bin/conda search --override-channels --channel iuc --channel conda-forge --channel bioconda --channel defaults '*seqt*'
Loading channels: done
# Name Version Build Channel
bioconductor-htseqtools 1.26.0 r3.4.1_0 bioconda
bioconductor-seqtools 1.10.0 r3.3.2_0 bioconda
bioconductor-seqtools 1.10.0 r3.4.1_0 bioconda
bioconductor-seqtools 1.12.0 r3.4.1_0 bioconda
seqtk r75 0 bioconda
seqtk r82 0 bioconda
seqtk r82 1 bioconda
seqtk r93 0 bioconda
seqtk 1.2 0 bioconda
seqtk 1.2 1 bioconda
Note
The Planemo command conda_search is a light wrapper around the underlying
conda search command but configured to use the same channels and other options as
Planemo and Galaxy. The following Conda command would also work to search:
$ $HOME/miniconda3/bin/conda -c iuc -c conda-forge -c bioconda '*seqt*'
For Conda versions 4.3.X or less, the search invocation would be something a bit different:
$ $HOME/miniconda3/bin/conda -c iuc -c conda-forge -c bioconda seqt
Alternatively the Anaconda website can be used to search for packages. Typing seqtk
into the search form on that page and clicking the top result will bring on to this page with information about the Bioconda package.
When using the website to search though, you need to aware of what channel you are using. By default, Planemo and Galaxy will search a few different Conda channels. While it is possible to configure a local Planemo or Galaxy to target different channels - the current best practice is to add tools to the existing channels.
The existing channels include:
Exercise - Leveraging Bioconda
Use the project_init command to download this exercise.
$ planemo project_init --template conda_exercises conda_exercises
$ cd conda_exercises/exercise_1
$ ls
pear.xml test-data
This project template contains a few exercises. The first uses an adapted
version of an IUC tool for PEAR - Paired-End reAd mergeR. This tool however has
no requirement tags and so will not work properly.
Run
planemo test pear.xmlto verify the tool does not function without dependencies defined.Use
--conda_requirementsflag withplanemo lintto verify it does indeed lack requirements.Use
planemo conda_searchor the Anaconda website to search for the correct package and version in a best practice channel.Update
pear.xmlwith the correctrequirementtags.Re-run the
lintcommand from above to verify the tool now has the correct dependency definition.Re-run the
testcommand from above to verify the tool test now works properly.
Building New Conda Packages
Frequently packages your tool will require are not found in Bioconda or conda-forge yet. In these cases, it is likely best to contribute your package to one of these projects. Unless the tool is exceedingly general Bioconda is usually the correct starting point.
Note
Many things that are not strictly or even remotely “bio” have been accepted into Bioconda - including tools for image analysis, natural language processing, and cheminformatics.
To get quickly learn to write Conda recipes for typical Galaxy tools, please read the following pieces of external documentation.
Contributing to Bioconda in particular focusing on
Contributing a recipe (through “Write a Recipe”)
Building conda packages in particular
Building conda packages with conda skeleton (the best approach for common scripting languages such as R and Python)
Then return to the Bioconda documentation and read
The rest of “Contributing a recipe” continuing from Testing locally
And finally Guidelines for bioconda recipes
These guidelines in particular can be skimmed depending on your recipe type, for instance that document provides specific advice for:
To go a little deeper, you may want to read:
And finally to debug problems the Bioconda troubleshooting documentation may prove useful.
Exercise - Build a Recipe
If you have just completed the exercise above - this exercise can be found in parent folder. Get
there with cd ../exercise_2. If not, the exercise can be downloaded with
$ planemo project_init --template conda_exercises conda_exercises
$ cd conda_exercises/exercise_2
$ ls
fleeqtk_seq.xml test-data
This is the skeleton of a tool wrapping the parody bioinformatics software package fleeqtk.
fleeqtk is a fork of the project seqtk that many Planemo tutorials are built around and the
example tool should hopefully be fairly familiar. fleeqtk version 1.3 can be downloaded
from here and built using
make. The result of make includes a single executable fleeqtk.
Clone and branch Bioconda.
Build a recipe for fleeqtk version 1.3. You may wish to start from scratch (
conda skeletonis not available for C programs like fleeqtk), or copy the recipe of seqtk and modify it for fleeqtk.Use
conda buildor Bioconda tooling to build the recipe.Run
planemo test --conda_use_local fleeqtk_seq.xmlto verify the resulting package works as expected.
Congratulations on writing a Conda recipe and building a package! Upon succesfully building and testing such a Bioconda package, you would normally push your branch to Github and open a pull request. This step is skipped here as to not pollute Bioconda with unneeded software packages.
Dependencies and Containers
For years Galaxy has supported running tools inside containers. The details of how to run Galaxy tools inside of containers varies depending on the Galaxy job runner but details can be found in Galaxy’s job_conf.xml sample file.
This document doesn’t describe how to run the containers, it describes how Galaxy
figures out which container to run for a given tool. There are currently
two strategies for finding containers for a tool - and they are each
discussed in detail in this document. The newer approach is a bit more experimental
but should be considered the best practice approach - it is
to allow Galaxy to find or build a BioContainers container using requirement
tags that resolve to best-practice Conda channels. The older approach is
to explicitly declare a container identifier in the tool XML.
While not as flexible as resolving arbitrary image IDs from URLs, the newer approach has a few key advantages that make them a best practice:
They provide superior reproducibility across Galaxy instances because the same binary Conda packages will automatically be used for both bare metal dependencies and inside containers.
They are constructed automatically from existing Conda packages so tool developers shouldn’t need to write
Dockerfiles or register projects on Docker Hub.They are produced using mulled which produce very small containers that make deployment easy.
Annotating
requirementtags reduces the opaqueness of the Docker process. With this method it is entirely traceable how the container was constructed from what sources were fetched, which exact build of every dependency was used, to how packages in the container were built. Beyond that metadata about the packages can be fetched from Bioconda (e.g. this).
Read more about this reproducibility stack in our preprint Practical computational reproducibility in the life sciences.
BioContainers
Note
This section is a continuation of Dependencies and Conda, please review that section for background information on resolving requirements with Conda.
Finding BioContainers
If a tool contains requirements in best practice Conda channels, a BioContainers-style container can be found or built for it.
As reminder, planemo lint --conda_requirements <tool.xml> can be used
to check if a tool contains only best-practice requirement tags. The lint
command can also be fed the --biocontainers flag to check if a
BioContainers container has been registered that is compatible with that tool.
Below is an example of using this with the completed seqtk_seq.xml
tool from the introductory tutorial.
$ planemo lint --biocontainers seqtk_seq.xml
Linting tool /home/planemo/workspace/planemo/project_templates/seqtk_complete/seqtk_seq.xml
Applying linter tests... CHECK
.. CHECK: 1 test(s) found.
Applying linter output... CHECK
.. INFO: 1 outputs found.
Applying linter inputs... CHECK
.. INFO: Found 9 input parameters.
Applying linter help... CHECK
.. CHECK: Tool contains help section.
.. CHECK: Help contains valid reStructuredText.
Applying linter general... CHECK
.. CHECK: Tool defines a version [0.1.0].
.. CHECK: Tool defines a name [Convert to FASTA (seqtk)].
.. CHECK: Tool defines an id [seqtk_seq].
.. CHECK: Tool targets 16.01 Galaxy profile.
Applying linter command... CHECK
.. INFO: Tool contains a command.
Applying linter citations... CHECK
.. CHECK: Found 1 likely valid citations.
Applying linter tool_xsd... CHECK
.. INFO: File validates against XML schema.
Applying linter biocontainer_registered... CHECK
.. INFO: BioContainer best-practice container found [quay.io/biocontainers/seqtk:1.2--0].
This last linter indicates that indeed a container has been registered
that is compatible with this tool – quay.io/biocontainers/seqtk:1.2--1.
We didn’t do any extra work to build this container for this tool, all
Bioconda recipes are packaged into containers and registered on quay.io
as part of the BioContainers project.
This tool can be tested using planemo test in its BioContainer
Docker container using the flag --biocontainers as shown below.
$ planemo test --biocontainers seqtk_seq.xml
...
2017-03-01 08:18:19,669 INFO [galaxy.tools.actions] Handled output named output1 for tool seqtk_seq (22.145 ms)
2017-03-01 08:18:19,683 INFO [galaxy.tools.actions] Added output datasets to history (14.604 ms)
2017-03-01 08:18:19,703 INFO [galaxy.tools.actions] Verified access to datasets for Job[unflushed,tool_id=seqtk_seq] (8.687 ms)
2017-03-01 08:18:19,704 INFO [galaxy.tools.actions] Setup for job Job[unflushed,tool_id=seqtk_seq] complete, ready to flush (20.380 ms)
2017-03-01 08:18:19,719 INFO [galaxy.tools.actions] Flushed transaction for job Job[id=2,tool_id=seqtk_seq] (15.191 ms)
2017-03-01 08:18:20,120 INFO [galaxy.jobs.handler] (2) Job dispatched
2017-03-01 08:18:20,311 DEBUG [galaxy.tools.deps] Using dependency seqtk version 1.2 of type conda
2017-03-01 08:18:20,312 DEBUG [galaxy.tools.deps] Using dependency seqtk version 1.2 of type conda
2017-03-01 08:18:20,325 INFO [galaxy.tools.deps.containers] Checking with container resolver [ExplicitDockerContainerResolver[]] found description [None]
2017-03-01 08:18:20,468 INFO [galaxy.tools.deps.containers] Checking with container resolver [CachedMulledDockerContainerResolver[namespace=None]] found description [None]
2017-03-01 08:18:20,881 INFO [galaxy.tools.deps.containers] Checking with container resolver [MulledDockerContainerResolver[namespace=biocontainers]] found description [ContainerDescription[identifier=quay.io/biocontainers/seqtk:1.2--0,type=docker]]
2017-03-01 08:18:20,904 INFO [galaxy.jobs.command_factory] Built script [/tmp/tmpw8_UQm/job_working_directory/000/2/tool_script.sh] for tool command [seqtk seq -A '/tmp/tmpw8_UQm/files/000/dataset_1.dat' > '/tmp/tmpw8_UQm/files/000/dataset_2.dat']
2017-03-01 08:18:21,060 DEBUG [galaxy.tools.deps] Using dependency samtools version None of type conda
2017-03-01 08:18:21,061 DEBUG [galaxy.tools.deps] Using dependency samtools version None of type conda
ok
----------------------------------------------------------------------
XML: /private/tmp/tmpw8_UQm/xunit.xml
----------------------------------------------------------------------
Ran 1 test in 11.926s
OK
2017-03-01 08:18:26,726 INFO [test_driver] Shutting down
...
2017-03-01 08:18:26,732 INFO [galaxy.jobs.handler] job handler stop queue stopped
Testing complete. HTML report is in "/home/planemo/workspace/planemo/tool_test_output.html".
All 1 test(s) executed passed.
seqtk_seq[0]: passed
$
A very important line here is:
2017-03-01 08:18:20,881 INFO [galaxy.tools.deps.containers] Checking with container resolver [MulledDockerContainerResolver[namespace=biocontainers]] found description [ContainerDescription[identifier=quay.io/biocontainers/seqtk:1.2--0,type=docker]]
This line indicates that Galaxy was able to find a container for this tool and executed the tool in that container.
For interactive testing, the planemo serve command also implements the
--biocontainers flag.
Building BioContainers
In this seqtk example the relevant BioContainer already existed on quay.io,
this won’t always be the case. For tools that contain multiple requirement
tags an existing container likely won’t exist. The mulled toolkit
(distributed with planemo or available standalone) can be used to build
containers for such tools. For such tools, if Galaxy is configured to use
BioContainers it will attempt to build these containers on the fly by default
(though this behavior can be disabled).
You can try it directly using the mull command in Planemo. The conda_testing
Planemo project template has a toy example tool with two requirements for
demonstrating this - bwa_and_samtools.xml.
$ planemo project_init --template=conda_testing conda_testing
$ cd conda_testing/
$ planemo mull bwa_and_samtools.xml
/Users/john/.planemo/involucro -v=3 -f /Users/john/workspace/planemo/.venv/lib/python2.7/site-packages/galaxy_lib-17.9.0-py2.7.egg/galaxy/tools/deps/mulled/invfile.lua -set CHANNELS='iuc,conda-forge,bioconda,defaults' -set TEST='true' -set TARGETS='samtools=1.3.1,bwa=0.7.15' -set REPO='quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820' -set BINDS='build/dist:/usr/local/' -set PREINSTALL='conda install --quiet --yes conda=4.3' build
/Users/john/.planemo/involucro -v=3 -f /Users/john/workspace/planemo/.venv/lib/python2.7/site-packages/galaxy_lib-17.9.0-py2.7.egg/galaxy/tools/deps/mulled/invfile.lua -set CHANNELS='iuc,conda-forge,bioconda,defaults' -set TEST='true' -set TARGETS='samtools=1.3.1,bwa=0.7.15' -set REPO='quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820' -set BINDS='build/dist:/usr/local/' -set PREINSTALL='conda install --quiet --yes conda=4.3' build
[Jun 19 11:28:35] DEBU Run file [/Users/john/workspace/planemo/.venv/lib/python2.7/site-packages/galaxy_lib-17.9.0-py2.7.egg/galaxy/tools/deps/mulled/invfile.lua]
[Jun 19 11:28:35] STEP Run image [continuumio/miniconda:latest] with command [[rm -rf /data/dist]]
[Jun 19 11:28:35] DEBU Creating container [step-730a02d79e]
[Jun 19 11:28:35] DEBU Created container [5e4b5f83c455 step-730a02d79e], starting it
[Jun 19 11:28:35] DEBU Container [5e4b5f83c455 step-730a02d79e] started, waiting for completion
[Jun 19 11:28:36] DEBU Container [5e4b5f83c455 step-730a02d79e] completed with exit code [0] as expected
[Jun 19 11:28:36] DEBU Container [5e4b5f83c455 step-730a02d79e] removed
[Jun 19 11:28:36] STEP Run image [continuumio/miniconda:latest] with command [[/bin/sh -c conda install --quiet --yes conda=4.3 && conda install -c iuc -c conda-forge -c bioconda -c defaults samtools=1.3.1 bwa=0.7.15 -p /usr/local --copy --yes --quiet]]
[Jun 19 11:28:36] DEBU Creating container [step-e95bf001c8]
[Jun 19 11:28:36] DEBU Created container [72b9ca0e56f8 step-e95bf001c8], starting it
[Jun 19 11:28:37] DEBU Container [72b9ca0e56f8 step-e95bf001c8] started, waiting for completion
[Jun 19 11:28:46] SOUT Fetching package metadata .........
[Jun 19 11:28:47] SOUT Solving package specifications: .
[Jun 19 11:28:50] SOUT
[Jun 19 11:28:50] SOUT Package plan for installation in environment /opt/conda:
[Jun 19 11:28:50] SOUT
[Jun 19 11:28:50] SOUT The following packages will be UPDATED:
[Jun 19 11:28:50] SOUT
[Jun 19 11:28:50] SOUT conda: 4.3.11-py27_0 --> 4.3.22-py27_0
[Jun 19 11:28:50] SOUT
[Jun 19 11:29:04] SOUT Fetching package metadata .................
[Jun 19 11:29:06] SOUT Solving package specifications: .
[Jun 19 11:29:56] SOUT
[Jun 19 11:29:56] SOUT Package plan for installation in environment /usr/local:
[Jun 19 11:29:56] SOUT
[Jun 19 11:29:56] SOUT The following NEW packages will be INSTALLED:
[Jun 19 11:29:56] SOUT
[Jun 19 11:29:56] SOUT bwa: 0.7.15-1 bioconda
[Jun 19 11:29:56] SOUT curl: 7.52.1-0
[Jun 19 11:29:56] SOUT libgcc: 5.2.0-0
[Jun 19 11:29:56] SOUT openssl: 1.0.2l-0
[Jun 19 11:29:56] SOUT pip: 9.0.1-py27_1
[Jun 19 11:29:56] SOUT python: 2.7.13-0
[Jun 19 11:29:56] SOUT readline: 6.2-2
[Jun 19 11:29:56] SOUT samtools: 1.3.1-5 bioconda
[Jun 19 11:29:56] SOUT setuptools: 27.2.0-py27_0
[Jun 19 11:29:56] SOUT sqlite: 3.13.0-0
[Jun 19 11:29:56] SOUT tk: 8.5.18-0
[Jun 19 11:29:56] SOUT wheel: 0.29.0-py27_0
[Jun 19 11:29:56] SOUT zlib: 1.2.8-3
[Jun 19 11:29:56] SOUT
[Jun 19 11:29:57] DEBU Container [72b9ca0e56f8 step-e95bf001c8] completed with exit code [0] as expected
[Jun 19 11:29:57] DEBU Container [72b9ca0e56f8 step-e95bf001c8] removed
[Jun 19 11:29:57] STEP Wrap [build/dist] as [quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0]
[Jun 19 11:29:57] DEBU Creating container [step-6f1c176372]
[Jun 19 11:29:58] DEBU Packing succeeded
As the output indicates, this command built the container named
quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0.
This is the same namespace / URL that would be used if or when published by
the BioContainers project.
Note
The first part of this mulled-v2 hash is a hash of the package names
that went into it, the second the packages used and build number. Check out
the Multi-package Containers
web application to explore best practice channels and build such hashes.
We can see this new container when running the Docker command images and
explore the new container interactively with docker run.
$ docker images
REPOSITORY TAG IMAGE ID CREATED SIZE
quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40 03dc1d2818d9de56938078b8b78b82d967c1f820-0 a740fe1e6a9e 16 hours ago 104 MB
quay.io/biocontainers/seqtk 1.2--0 10bc359ebd30 2 days ago 7.34 MB
continuumio/miniconda latest 6965a4889098 3 weeks ago 437 MB
bgruening/busybox-bash 0.1 3d974f51245c 9 months ago 6.73 MB
$ docker run -i -t quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0 /bin/bash
bash-4.2# which samtools
/usr/local/bin/samtools
bash-4.2# which bwa
/usr/local/bin/bwa
As before, we can test running the tool inside its container in Galaxy using
the --biocontainers flag.
$ planemo test --biocontainers bwa_and_samtools.xml
...
2017-03-01 10:20:58,077 INFO [galaxy.tools.actions] Handled output named output_2 for tool bwa_and_samtools (17.443 ms)
2017-03-01 10:20:58,090 INFO [galaxy.tools.actions] Added output datasets to history (12.935 ms)
2017-03-01 10:20:58,095 INFO [galaxy.tools.actions] Verified access to datasets for Job[unflushed,tool_id=bwa_and_samtools] (0.021 ms)
2017-03-01 10:20:58,096 INFO [galaxy.tools.actions] Setup for job Job[unflushed,tool_id=bwa_and_samtools] complete, ready to flush (5.755 ms)
2017-03-01 10:20:58,116 INFO [galaxy.tools.actions] Flushed transaction for job Job[id=1,tool_id=bwa_and_samtools] (19.582 ms)
2017-03-01 10:20:58,869 INFO [galaxy.jobs.handler] (1) Job dispatched
2017-03-01 10:20:59,067 DEBUG [galaxy.tools.deps] Using dependency bwa version 0.7.15 of type conda
2017-03-01 10:20:59,067 DEBUG [galaxy.tools.deps] Using dependency samtools version 1.3.1 of type conda
2017-03-01 10:20:59,067 DEBUG [galaxy.tools.deps] Using dependency bwa version 0.7.15 of type conda
2017-03-01 10:20:59,068 DEBUG [galaxy.tools.deps] Using dependency samtools version 1.3.1 of type conda
2017-03-01 10:20:59,083 INFO [galaxy.tools.deps.containers] Checking with container resolver [ExplicitContainerResolver[]] found description [None]
2017-03-01 10:20:59,142 INFO [galaxy.tools.deps.containers] Checking with container resolver [CachedMulledDockerContainerResolver[namespace=biocontainers]] found description [ContainerDescription[identifier=quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0,type=docker]]
2017-03-01 10:20:59,163 INFO [galaxy.jobs.command_factory] Built script [/tmp/tmpQs0gyp/job_working_directory/000/1/tool_script.sh] for tool command [bwa > /tmp/tmpQs0gyp/files/000/dataset_1.dat 2>&1 ; samtools > /tmp/tmpQs0gyp/files/000/dataset_2.dat 2>&1]
2017-03-01 10:20:59,367 DEBUG [galaxy.tools.deps] Using dependency samtools version None of type conda
2017-03-01 10:20:59,367 DEBUG [galaxy.tools.deps] Using dependency samtools version None of type conda
ok
----------------------------------------------------------------------
XML: /private/tmp/tmpQs0gyp/xunit.xml
----------------------------------------------------------------------
Ran 1 test in 7.553s
OK
2017-03-01 10:21:05,223 INFO [test_driver] Shutting down
2017-03-01 10:21:05,224 INFO [test_driver] Shutting down embedded galaxy web server
2017-03-01 10:21:05,226 INFO [test_driver] Embedded web server galaxy stopped
2017-03-01 10:21:05,226 INFO [test_driver] Stopping application galaxy
...
2017-03-01 10:21:05,228 INFO [galaxy.jobs.handler] job handler stop queue stopped
Testing complete. HTML report is in "/home/planemo/workspace/planemo/tool_test_output.html".
All 1 test(s) executed passed.
bwa_and_samtools[0]: passed
In particular take note of the line:
2017-03-01 10:20:59,142 INFO [galaxy.tools.deps.containers] Checking with container resolver [CachedMulledDockerContainerResolver[namespace=biocontainers]] found description [ContainerDescription[identifier=quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0,type=docker]]
Here we can see the container ID (quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0)
from earlier has been cached on our Docker host is picked up by Galaxy. This is used to run the simple
tool tests and indeed they pass.
In our initial seqtk example, the container resolver that matched was of type
MulledDockerContainerResolver indicating that the Docker image would be downloaded
from the BioContainer repository and this time the resolve that matched was of type
CachedMulledDockerContainerResolver meaning that Galaxy would just use the locally
cached version from the Docker host (i.e. the one we built with planemo mull
above).
Note
Planemo doesn’t yet expose options that make it possible to build mulled
containers for local packages that have yet to be published to anaconda.org
but the mulled toolkit allows this. See mulled documentation for more
information. However, once a container for a local package is built with
mulled-build-tool the --biocontainers command should work to test
it.
Publishing BioContainers
Building unpublished BioContainers on the fly is great for testing but for production use and to increase reproducibility such containers should ideally be published as well.
BioContainers maintains a registry of package combinations to be published
using these long mulled hashes. This registry is represented as a Github repository
named multi-package-containers.
The Planemo command container_register will inspect a tool and open a
Github pull request to add the tool’s combination
of packages to the registry. Once merged, this pull request will
result in the corresponding BioContainers image to be published (with the
correct mulled has as its name) - these can be subsequently be picked up by
Galaxy.
Various Github related settings need to be configured in order for Planemo
to be able to open pull requests on your behalf as part of the
container_register command. To simplify all of this - the Planemo community
maintains a list of Github repositories containing Galaxy and/or CWL tools that
are scanned daily by Travis. For each such repository, the Travis job will run
container_register across the repository on all tools resulting in new registry
pull requests for all new combinations of tools. This list is maintained
in a script named monitor.sh in the planemo-monitor repository. The easiest way
to ensure new containers are built for your tools is simply to open open a pull
request to add your tool repositories to this list.
Explicit Annotation
This section of documentation needs to be filled out but a detailed example is worked through this documentation from Aaron Petkau (@apetkau) built at the 2014 Galaxy Community Conference Hackathon.