Building Galaxy Tools (Using the Planemo Appliance)
This tutorial is a gentle introduction to writing Galaxy tools using the Planemo virtual appliance (available as OVA, Docker and Vagrant). Check out these instructions for obtaining the virtual appliance if you have not done so already.
Note
Please note that you can leverage the clipboard for sharing text between the
virtual image environment and your host system. To copy in the VM terminal use
ctrl + shift + C and to paste use ctrl + shift + V. To copy
in the VM Firefox browser use ctrl + C.
Use the corresponding commands on your host system (e.g. Command + C on MacOS).
The Basics
This guide is going to demonstrate building up tools for commands from Heng Li’s Seqtk package - a package for processing sequence data in FASTA and FASTQ files.
To get started let’s install Seqtk. Here we are going to use conda to
install Seqtk - but however you obtain it should be fine.
$ conda install --force --yes -c conda-forge -c bioconda seqtk=1.2
... seqtk installation ...
$ seqtk seq
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
Next we will download an example FASTQ file and test out the a simple Seqtk
command - seq which converts FASTQ files into FASTA.
$ wget https://raw.githubusercontent.com/galaxyproject/galaxy-test-data/master/2.fastq
$ seqtk seq -A 2.fastq > 2.fasta
$ cat 2.fasta
>EAS54_6_R1_2_1_413_324
CCCTTCTTGTCTTCAGCGTTTCTCC
>EAS54_6_R1_2_1_540_792
TTGGCAGGCCAAGGCCGATGGATCA
>EAS54_6_R1_2_1_443_348
GTTGCTTCTGGCGTGGGTGGGGGGG
For fully featured Seqtk wrappers check out Helena Rasche’s wrappers on GitHub.
Galaxy tool files are just XML files, so at this point one could
open a text editor and start writing the tool. Planemo has a command
tool_init to quickly generate some of the boilerplate XML, so let’s
start by doing that.
$ planemo tool_init --id 'seqtk_seq' --name 'Convert to FASTA (seqtk)'
The tool_init command can take various complex arguments - but the two
most basic ones are shown above --id and --name. Every Galaxy tool
needs an id (this is a short identifier used by Galaxy itself to identify the
tool) and a name (this is displayed to the Galaxy user and should be a short
description of the tool). A tool’s name can have whitespace but its id
must not.
The above command will generate the file seqtk_seq.xml - which looks
like this.
<tool id="seqtk_seq" name="Convert to FASTA (seqtk)" version="0.1.0">
<requirements>
</requirements>
<command detect_errors="exit_code"><![CDATA[
TODO: Fill in command template.
]]></command>
<inputs>
</inputs>
<outputs>
</outputs>
<help><![CDATA[
TODO: Fill in help.
]]></help>
</tool>
This tool file has the common sections required for a Galaxy tool but you will still need to open up the editor and fill out the command template, describe input parameters, tool outputs, write a help section, etc.
The tool_init command can do a little bit better than this as well. We can
use the test command we tried above seqtk seq -A 2.fastq > 2.fasta as
an example to generate a command block by specifing the inputs and the outputs
as follows.
$ planemo tool_init --force \
--id 'seqtk_seq' \
--name 'Convert to FASTA (seqtk)' \
--requirement seqtk@1.2 \
--example_command 'seqtk seq -A 2.fastq > 2.fasta' \
--example_input 2.fastq \
--example_output 2.fasta
This will generate the following XML file - which now has correct definitions for the input and output as well as an actual command template.
<tool id="seqtk_seq" name="Convert to FASTA (seqtk)" version="0.1.0">
<requirements>
<requirement type="package" version="1.2">seqtk</requirement>
</requirements>
<command detect_errors="exit_code"><![CDATA[
seqtk seq -A '$input1' > '$output1'
]]></command>
<inputs>
<param type="data" name="input1" format="fastq" />
</inputs>
<outputs>
<data name="output1" format="fasta" />
</outputs>
<help><![CDATA[
TODO: Fill in help.
]]></help>
</tool>
As shown at the beginning of this section, the command seqtk seq generates
a help message for the seq command. tool_init can take that help message and
stick it right in the generated tool file using the help_from_command option.
Generally command help messages aren’t exactly appropriate for tools since they mention argument names and simillar details that are abstracted away by the tool - but they can be an excellent place to start.
The following Planemo’s tool_init call has been enhanced to use --help_from_command.
$ planemo tool_init --force \
--id 'seqtk_seq' \
--name 'Convert to FASTA (seqtk)' \
--requirement seqtk@1.2 \
--example_command 'seqtk seq -A 2.fastq > 2.fasta' \
--example_input 2.fastq \
--example_output 2.fasta \
--test_case \
--cite_url 'https://github.com/lh3/seqtk' \
--help_from_command 'seqtk seq'
In addition to demonstrating --help_from_command, this demonstrates generating
a test case from our example with --test_case and adding a citation for the
underlying tool. The resulting tool XML file is:
<tool id="seqtk_seq" name="Convert to FASTA (seqtk)" version="0.1.0">
<requirements>
<requirement type="package" version="1.2">seqtk</requirement>
</requirements>
<command detect_errors="exit_code"><![CDATA[
seqtk seq -A '$input1' > '$output1'
]]></command>
<inputs>
<param type="data" name="input1" format="fastq" />
</inputs>
<outputs>
<data name="output1" format="fasta" />
</outputs>
<tests>
<test>
<param name="input1" value="2.fastq"/>
<output name="output1" file="2.fasta"/>
</test>
</tests>
<help><![CDATA[
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
-U convert all bases to uppercases
-S strip of white spaces in sequences
]]></help>
<citations>
<citation type="bibtex">
@misc{githubseqtk,
author = {LastTODO, FirstTODO},
year = {TODO},
title = {seqtk},
publisher = {GitHub},
journal = {GitHub repository},
url = {https://github.com/lh3/seqtk},
}</citation>
</citations>
</tool>
At this point we have a fairly a functional Galaxy tool with test and help. This was
a pretty simple example - usually you will need to put more work into the tool
to get to this point - tool_init is really just designed to get you
started.
Now lets lint and test the tool we have developed. The Planemo’s lint (or
just l) command will review tool for XML validity, obvious mistakes, and
compliance with IUC best practices.
$ planemo l
Linting tool /opt/galaxy/tools/seqtk_seq.xml
Applying linter tests... CHECK
.. CHECK: 1 test(s) found.
Applying linter output... CHECK
.. INFO: 1 outputs found.
Applying linter inputs... CHECK
.. INFO: Found 1 input parameters.
Applying linter help... CHECK
.. CHECK: Tool contains help section.
.. CHECK: Help contains valid reStructuredText.
Applying linter general... CHECK
.. CHECK: Tool defines a version [0.1.0].
.. CHECK: Tool defines a name [Convert to FASTA (seqtk)].
.. CHECK: Tool defines an id [seqtk_seq].
Applying linter command... CHECK
.. INFO: Tool contains a command.
Applying linter citations... CHECK
.. CHECK: Found 1 likely valid citations.
By default lint will find all the tools in your current working directory,
but we could have specified a particular tool with planemo lint
seqtk_seq.xml.
Next we can run our tool’s functional test with the test (or just t)
command. This will print a lot of output (as it starts a Galaxy instance) but should
ultimately reveal our one test passed.
$ planemo t
... Galaxy starts and runs the test ...
All 1 test(s) executed passed.
seqtk_seq[0]: passed
You can use the following command to open up the test results in your browser.
$ firefox /opt/galaxy/tools/tool_test_output.html
Normally planemo requires an existing Galaxy instance to point at to run
the t (or test command) - but the virtual appliance has a
Galaxy instance preconfigured and registered with planemo.
Simple Parameters
We have built a tool wrapper for the seqtk seq command - but this tool
actually has additional options that we may wish to expose the Galaxy user.
Lets take a few of the parameters from the help command and build Galaxy
param blocks to stick in the tool’s inputs block.
-V shift quality by '(-Q) - 33'
In the previous section we saw param block of type data for input
files, but there are many different kinds of parameters one can use.
Flag parameters such as the above -V parameter are frequently
represented by boolean parameters in Galaxy tool XML.
<param name="shift_quality" type="boolean" label="Shift quality"
truevalue="-V" falsevalue=""
help="shift quality by '(-Q) - 33' (-V)" />
We can then stick $shift_quality in our command block and if the user
has selected this option it will be expanded as -V (since we have defined
this as the truevalue). If the user hasn’t selected this option
$shift_quality will just expand as an empty string and not affect the
generated command line.
Now consider the following seqtk seq parameters:
-q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
These can be translated into Galaxy parameters as:
<param name="quality_min" type="integer" label="Mask bases with quality lower than"
value="0" min="0" max="255" help="(-q)" />
<param name="quality_max" type="integer" label="Mask bases with quality higher than"
value="255" min="0" max="255" help="(-X)" />
These can be add to the command tag as -q $quality_min -X $quality_max.
At this point the tool would look like:
<tool id="seqtk_seq" name="Convert to FASTA (seqtk)" version="0.1.0">
<requirements>
<requirement type="package" version="1.2">seqtk</requirement>
</requirements>
<command detect_errors="exit_code"><![CDATA[
seqtk seq
$shift_quality
-q $quality_min
-X $quality_max
-a '$input1' > '$output1'
]]></command>
<inputs>
<param type="data" name="input1" format="fastq" />
<param name="shift_quality" type="boolean" label="Shift quality"
truevalue="-V" falsevalue=""
help="shift quality by '(-Q) - 33' (-V)" />
<param name="quality_min" type="integer" label="Mask bases with quality lower than"
value="0" min="0" max="255" help="(-q)" />
<param name="quality_max" type="integer" label="Mask bases with quality higher than"
value="255" min="0" max="255" help="(-X)" />
</inputs>
<outputs>
<data name="output1" format="fasta" />
</outputs>
<tests>
<test>
<param name="input1" value="2.fastq"/>
<output name="output1" file="2.fasta"/>
</test>
</tests>
<help><![CDATA[
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
-U convert all bases to uppercases
]]></help>
<citations>
<citation type="bibtex">
@misc{githubseqtk,
author = {LastTODO, FirstTODO},
year = {TODO},
title = {seqtk},
publisher = {GitHub},
journal = {GitHub repository},
url = {https://github.com/lh3/seqtk},
}</citation>
</citations>
</tool>
Conditional Parameters
The previous parameters were simple because they always appeared, now consider.
-M FILE mask regions in BED or name list FILE [null]
We can mark this data type param as optional by adding the attribute
optional="true".
<param name="mask_regions" type="data" label="Mask regions in BED"
format="bed" help="(-M)" optional="true" />
Then instead of just using $mask_regions directly in the command
block, one can wrap it in an if statement (because tool XML files
support Cheetah).
#if $mask_regions
-M '$mask_regions'
#end if
Next consider the parameters:
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
In this case, the -s random seed parameter should only be seen or used if
the sample parameter is set. We can express this using a conditional
block.
<conditional name="sample">
<param name="sample_selector" type="boolean" label="Sample fraction of sequences" />
<when value="true">
<param name="fraction" label="Fraction" type="float" value="1.0"
help="(-f)" />
<param name="seed" label="Random seed" type="integer" value="11"
help="(-s)" />
</when>
<when value="false">
</when>
</conditional>
In our command block, we can again use an if statement to include these
parameters.
#if $sample.sample_selector
-f $sample.fraction -s $sample.seed
#end if
Notice we must reference the parameters using the sample. prefix since
they are defined within the sample conditional block.
The newest version of this tool is now
<tool id="seqtk_seq" name="Convert to FASTA (seqtk)" version="0.1.0">
<requirements>
<requirement type="package" version="1.2">seqtk</requirement>
</requirements>
<command detect_errors="exit_code"><![CDATA[
seqtk seq
$shift_quality
-q $quality_min
-X $quality_max
#if $mask_regions
-M '$mask_regions'
#end if
#if $sample.sample
-f $sample.fraction
-s $sample.seed
#end if
-a '$input1' > '$output1'
]]></command>
<inputs>
<param type="data" name="input1" format="fastq" />
<param name="shift_quality" type="boolean" label="Shift quality"
truevalue="-V" falsevalue=""
help="shift quality by '(-Q) - 33' (-V)" />
<param name="quality_min" type="integer" label="Mask bases with quality lower than"
value="0" min="0" max="255" help="(-q)" />
<param name="quality_max" type="integer" label="Mask bases with quality higher than"
value="255" min="0" max="255" help="(-X)" />
<param name="mask_regions" type="data" label="Mask regions in BED"
format="bed" help="(-M)" optional="true" />
<conditional name="sample">
<param name="sample" type="boolean" label="Sample fraction of sequences" />
<when value="true">
<param name="fraction" label="Fraction" type="float" value="1.0"
help="(-f)" />
<param name="seed" label="Random seed" type="integer" value="11"
help="(-s)" />
</when>
<when value="false">
</when>
</conditional>
</inputs>
<outputs>
<data name="output1" format="fasta" />
</outputs>
<tests>
<test>
<param name="input1" value="2.fastq"/>
<output name="output1" file="2.fasta"/>
</test>
</tests>
<help><![CDATA[
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
-U convert all bases to uppercases
]]></help>
<citations>
<citation type="bibtex">
@misc{githubseqtk,
author = {LastTODO, FirstTODO},
year = {TODO},
title = {seqtk},
publisher = {GitHub},
journal = {GitHub repository},
url = {https://github.com/lh3/seqtk},
}</citation>
</citations>
</tool>
For tools like this where there are many options but in the most uses the defaults
are preferred - a common idiom is to break the parameters into simple and
advanced sections using a conditional.
Updating this tool to use that idiom might look as follows.
<tool id="seqtk_seq" name="Convert to FASTA (seqtk)" version="0.1.0">
<requirements>
<requirement type="package" version="1.2">seqtk</requirement>
</requirements>
<command detect_errors="exit_code"><![CDATA[
seqtk seq
#if $settings.advanced == "advanced"
$settings.shift_quality
-q $settings.quality_min
-X $settings.quality_max
#if $settings.mask_regions
-M '$settings.mask_regions'
#end if
#if $settings.sample.sample
-f $settings.sample.fraction
-s $settings.sample.seed
#end if
#end if
-a '$input1' > '$output1'
]]></command>
<inputs>
<param type="data" name="input1" format="fastq" />
<conditional name="settings">
<param name="advanced" type="select" label="Specify advanced parameters">
<option value="simple" selected="true">No, use program defaults.</option>
<option value="advanced">Yes, see full parameter list.</option>
</param>
<when value="simple">
</when>
<when value="advanced">
<param name="shift_quality" type="boolean" label="Shift quality"
truevalue="-V" falsevalue=""
help="shift quality by '(-Q) - 33' (-V)" />
<param name="quality_min" type="integer" label="Mask bases with quality lower than"
value="0" min="0" max="255" help="(-q)" />
<param name="quality_max" type="integer" label="Mask bases with quality higher than"
value="255" min="0" max="255" help="(-X)" />
<param name="mask_regions" type="data" label="Mask regions in BED"
format="bed" help="(-M)" optional="true" />
<conditional name="sample">
<param name="sample" type="boolean" label="Sample fraction of sequences" />
<when value="true">
<param name="fraction" label="Fraction" type="float" value="1.0"
help="(-f)" />
<param name="seed" label="Random seed" type="integer" value="11"
help="(-s)" />
</when>
<when value="false">
</when>
</conditional>
</when>
</conditional>
</inputs>
<outputs>
<data name="output1" format="fasta" />
</outputs>
<tests>
<test>
<param name="input1" value="2.fastq"/>
<output name="output1" file="2.fasta"/>
</test>
</tests>
<help><![CDATA[
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
-U convert all bases to uppercases
]]></help>
<citations>
<citation type="bibtex">
@misc{githubseqtk,
author = {LastTODO, FirstTODO},
year = {TODO},
title = {seqtk},
publisher = {GitHub},
journal = {GitHub repository},
url = {https://github.com/lh3/seqtk},
}</citation>
</citations>
</tool>
Publishing to the Tool Shed
Now that the tool is working and useful - it is time to publish it to the Tool Shed. The Galaxy Tool Shed (referred to colloquially in Planemo as the “shed”) can store Galaxy tools, dependency definitions, and workflows among other Galaxy artifacts. Shed’s goal is to make it easy for any Galaxy to install these.
Configuring a Tool Shed Account
The planemo
appliance comes pre-configured with a local Tool Shed and Planemo is
configured to talk to it via ~/.planemo.yml configuration file.
Check out the publishing docs
for information on setting up this file on your development environment.
Creating a Repository
Planemo can be used to publish “repositories” to the Tool Shed. A
single GitHub repository or locally managed directory of tools may correspond
to any number of Tool Shed repositories. Planemo maps files to Tool Shed
repositories using a special file called .shed.yml.
From a directory containing tools the shed_init
command
can be used to bootstrap a new .shed.yml file.
$ planemo shed_init --name=seqtk_seq \
--owner=planemo \
--description=seqtk_seq \
--long_description="Tool that converts FASTQ to FASTA files using seqtk" \
--category="Fastq Manipulation"
The resulting .shed.yml file will look something like this:
categories: [Fastq Manipulation]
description: seqtk_seq
long_description: Tool that converts FASTQ to FASTA files using seqtk
name: seqtk_seq
owner: planemo
There is not a lot of magic happening here, this file could easily be created
directly with a text editor - but the command has a --help to assist you
and does some very basic validation. More information on .shed.yml
can be found as part of the IUC’s best practice documentation.
This configuration file and shed artifacts can be quickly linted using the following command.
$ planemo shed_lint --tools
Once the details in the .shed.yml are set and it is time to create the remote
repository and upload artifacts to it - the following two commands can be used
- the first only needs to be run once and creates the repository based on the
metadata in .shed.yml and the second uploads your actual artifacts to it.
$ planemo shed_create --shed_target local
Repository created
cd '/opt/galaxy/tools' && git rev-parse HEAD
Repository seqtk_seq updated successfully.
You can now navigate to the local shed (likely at http://localhost:9009/) and see the repository there.
Optionally you can login with username planemo@test.com
and password planemo but it is not necessary.
Updating a Repository
In order to push further changes in your local tool development directory
to the shed you would run the shed_update command as follows.
$ planemo shed_update --shed_target local
Serving a Tool from Shed
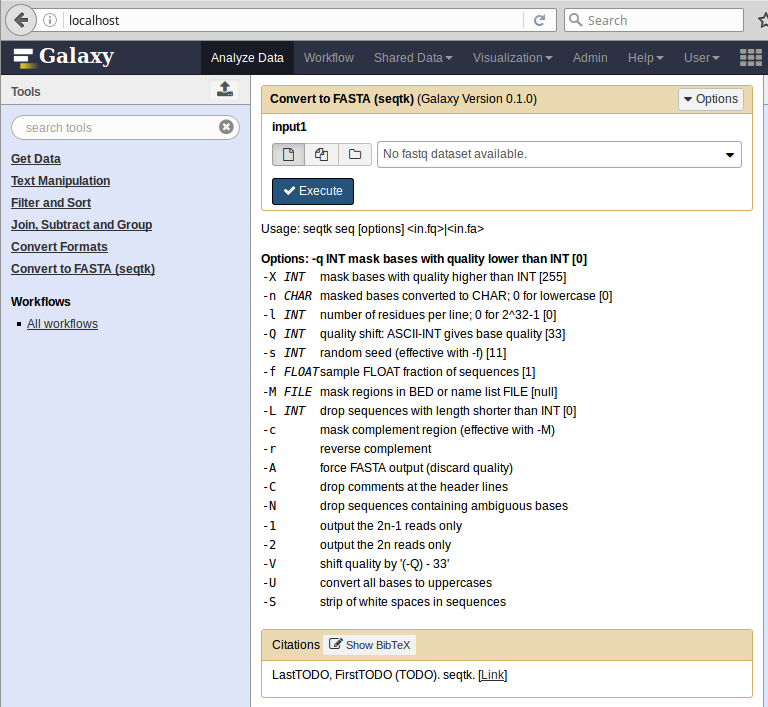
Once tools (and possible required dependency files) have been published, the whole thing can be automatically installed and the tool served in local Galaxy using this command.
$ planemo shed_serve --shed_target local
Note
During this tutorial we did not “teach” Galaxy how to obtain
the seqtk software so our tool (and thus Galaxy) just expects the command
seqtk to be available. The seqtk software here is a so called dependency
of our tool and in order for our tool to be fully installable we need to
create a “recipe” for Galaxy so it knows how to obtain it. This is covered
in other sections of this documentation as well as on the
wiki.
Main Tool Shed
Once your artifacts are ready for publication to the Main Tool Shed, the following command creates a repository there and populates it with your contents.
$ planemo shed_create --shed_target toolshed
The planemo machine isn’t preconfigured to allow publishing to the Main Tool Shed so this command will not work here. See the more complete publishing docs for full details about how to setup Planemo to publish to the Main and Test Tool Shed - the process is very similar.
Wrapping a Script
Many common bioinformatics applications are available on the Tool Shed already and so a common development task is to integrate scripts of various complexity into Galaxy.
Consider the following small Perl script.
#!/usr/bin/perl -w
# usage : perl toolExample.pl <FASTA file> <output file>
open (IN, "<$ARGV[0]");
open (OUT, ">$ARGV[1]");
while (<IN>) {
chop;
if (m/^>/) {
s/^>//;
if ($. > 1) {
print OUT sprintf("%.3f", $gc/$length) . "\n";
}
$gc = 0;
$length = 0;
} else {
++$gc while m/[gc]/ig;
$length += length $_;
}
}
print OUT sprintf("%.3f", $gc/$length) . "\n";
close( IN );
close( OUT );
One can build a tool for this script as follows and place the script in
the same directory as the tool XML file itself. The special value
$__tool_directory__ here refers to the directory your tool lives in.
<tool id="gc_content" name="Compute GC content">
<description>for each sequence in a file</description>
<command>perl '$__tool_directory__/gc_content.pl' '$input' output.tsv</command>
<inputs>
<param name="input" type="data" format="fasta" label="Source file"/>
</inputs>
<outputs>
<data name="output" format="tabular" from_work_dir="output.tsv" />
</outputs>
<help>
This tool computes GC content from a FASTA file.
</help>
</tool>
Macros
If your desire is to write a tool for a single relatively simple application or script - this section should be skipped. If you hope to maintain a collection of related tools - experience suggests you will realize there is a lot of duplicated XML to do this well. Galaxy tool XML macros can help reduce this duplication.
Planemo’s tool_init command can be used to generate a macro file
appropriate for suites of tools by using the --macros flag. Consider the
following variant of the previous tool_init command (the only difference
is now we are adding the --macros flag).
$ planemo tool_init --force \
--macros \
--id 'seqtk_seq' \
--name 'Convert to FASTA (seqtk)' \
--requirement seqtk@1.2 \
--example_command 'seqtk seq -A 2.fastq > 2.fasta' \
--example_input 2.fastq \
--example_output 2.fasta \
--test_case \
--help_from_command 'seqtk seq'
This will produce the two files in your current directory instead of just one
- seqtk_seq.xml and macros.xml.
<tool id="seqtk_seq" name="Convert to FASTA (seqtk)" version="0.1.0">
<macros>
<import>macros.xml</import>
</macros>
<expand macro="requirements" />
<command detect_errors="exit_code"><![CDATA[
seqtk seq -A '$input1' > '$output1'
]]></command>
<inputs>
<param type="data" name="input1" format="fastq" />
</inputs>
<outputs>
<data name="output1" format="fasta" />
</outputs>
<tests>
<test>
<param name="input1" value="2.fastq"/>
<output name="output1" file="2.fasta"/>
</test>
</tests>
<help><![CDATA[
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
]]></help>
<expand macro="citations" />
</tool>
<macros>
<xml name="requirements">
<requirements>
<requirement type="package" version="1.2">seqtk</requirement>
<yield/>
</requirements>
</xml>
<xml name="citations">
<citations>
<yield />
</citations>
</xml>
</macros>
As you can see in the above code macros are reusable chunks of XML that make it easier to avoid duplication and keep your XML concise.
Further reading:
Macros syntax on the Galaxy Wiki.
GATK tools (example tools making extensive use of macros)
gemini tools (example tools making extensive use of macros)
bedtools tools (example tools making extensive use of macros)
Macros implementation details - Pull Request #129 and Pull Request #140