Advanced Tool Development Topics
This tutorial covers some more advanced tool development topics. It assumes some basic knowledge about developing CWL tools and that you have an environment with Planemo available - check out the CWL User Guide CWL and the Planemo CWL intro tutorial if you have never developed a CWL tool.
Dependencies and Conda
Specifying and Using Software Requirements
Note
Planemo requires a Conda installation to target with its various Conda
related commands. A properly configured Conda installation can be initialized
with the conda_init command. This should only need to be executed once
per development machine.
$ planemo conda_init
galaxy.tools.deps.conda_util INFO: Installing conda, this may take several minutes.
wget -q --recursive -O /var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/conda_installLW5zn1.sh https://repo.continuum.io/miniconda/Miniconda3-4.3.31-MacOSX-x86_64.sh
bash /var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/conda_installLW5zn1.sh -b -p /Users/john/miniconda3
PREFIX=/Users/john/miniconda3
installing: python-3.6.3-h47c878a_7 ...
Python 3.6.3 :: Anaconda, Inc.
installing: ca-certificates-2017.08.26-ha1e5d58_0 ...
installing: conda-env-2.6.0-h36134e3_0 ...
installing: libcxxabi-4.0.1-hebd6815_0 ...
installing: tk-8.6.7-h35a86e2_3 ...
installing: xz-5.2.3-h0278029_2 ...
installing: yaml-0.1.7-hc338f04_2 ...
installing: zlib-1.2.11-hf3cbc9b_2 ...
installing: libcxx-4.0.1-h579ed51_0 ...
installing: openssl-1.0.2n-hdbc3d79_0 ...
installing: libffi-3.2.1-h475c297_4 ...
installing: ncurses-6.0-hd04f020_2 ...
installing: libedit-3.1-hb4e282d_0 ...
installing: readline-7.0-hc1231fa_4 ...
installing: sqlite-3.20.1-h7e4c145_2 ...
installing: asn1crypto-0.23.0-py36h782d450_0 ...
installing: certifi-2017.11.5-py36ha569be9_0 ...
installing: chardet-3.0.4-py36h96c241c_1 ...
installing: idna-2.6-py36h8628d0a_1 ...
installing: pycosat-0.6.3-py36hee92d8f_0 ...
installing: pycparser-2.18-py36h724b2fc_1 ...
installing: pysocks-1.6.7-py36hfa33cec_1 ...
installing: python.app-2-py36h54569d5_7 ...
installing: ruamel_yaml-0.11.14-py36h9d7ade0_2 ...
installing: six-1.11.0-py36h0e22d5e_1 ...
installing: cffi-1.11.2-py36hd3e6348_0 ...
installing: setuptools-36.5.0-py36h2134326_0 ...
installing: cryptography-2.1.4-py36h842514c_0 ...
installing: wheel-0.30.0-py36h5eb2c71_1 ...
installing: pip-9.0.1-py36h1555ced_4 ...
installing: pyopenssl-17.5.0-py36h51e4350_0 ...
installing: urllib3-1.22-py36h68b9469_0 ...
installing: requests-2.18.4-py36h4516966_1 ...
installing: conda-4.3.31-py36_0 ...
installation finished.
/Users/john/miniconda3/bin/conda install -y --override-channels --channel iuc --channel conda-forge --channel bioconda --channel defaults conda=4.3.33 conda-build=2.1.18
Fetching package metadata ...................
Solving package specifications: .
Package plan for installation in environment /Users/john/miniconda3:
The following NEW packages will be INSTALLED:
beautifulsoup4: 4.6.0-py36_0 conda-forge
conda-build: 2.1.18-py36_0 conda-forge
conda-verify: 2.0.0-py36_0 conda-forge
filelock: 3.0.4-py36_0 conda-forge
jinja2: 2.10-py36_0 conda-forge
markupsafe: 1.0-py36_0 conda-forge
pkginfo: 1.4.2-py36_0 conda-forge
pycrypto: 2.6.1-py36_1 conda-forge
pyyaml: 3.12-py36_1 conda-forge
The following packages will be UPDATED:
conda: 4.3.31-py36_0 --> 4.3.33-py36_0 conda-forge
beautifulsoup4 100% |###################################################################| Time: 0:00:00 782.08 kB/s
filelock-3.0.4 100% |###################################################################| Time: 0:00:00 7.95 MB/s
markupsafe-1.0 100% |###################################################################| Time: 0:00:00 5.82 MB/s
pkginfo-1.4.2- 100% |###################################################################| Time: 0:00:00 1.18 MB/s
pycrypto-2.6.1 100% |###################################################################| Time: 0:00:00 1.69 MB/s
pyyaml-3.12-py 100% |###################################################################| Time: 0:00:00 3.31 MB/s
conda-verify-2 100% |###################################################################| Time: 0:00:00 6.91 MB/s
jinja2-2.10-py 100% |###################################################################| Time: 0:00:00 2.81 MB/s
conda-4.3.33-p 100% |###################################################################| Time: 0:00:00 621.27 kB/s
conda-build-2. 100% |###################################################################| Time: 0:00:00 2.16 MB/s
Conda installation succeeded - Conda is available at '/Users/john/miniconda3/bin/conda'
Note
Why not just use containers?
Containers are great, use containers (be it Docker, Singularity, etc.) whenever possible to
increase reproducibility and portability of your tools and workflow. Building ad hoc containers
to support CWL tools (e.g. custom Dockerfile definitions) has serious limitations, in the next
tutorial on containers we will argue that using Biocontainers built or discovered
from your tool’s Software Requirements is a superior approach.
Besides leading to better containers, there are other reasons to describe Software Requirements also - it will allow your tool to be used in environments without container runtimes available and provides valuable and actionable metadata about the computation described by the tool.
Read more about this whole dependency stack in our preprint Practical computational reproducibility in the life sciences
The Common Workflow Language specification loosely describes Software Requirements - a way to map CWL hints to packages, environment modules, or any other mechanism to describe dependencies for running a tool outside of a container. The large and active Galaxy tool development community has built an open source library and set of best practices for describing dependencies for Galaxy that should work just as well for CWL. The library has been integrated with cwltool and Toil to enable CWL tool authors and users to leverage the power and flexibility of the Galaxy dependency management and best practices.
While Software Requirements can be configured to resolve dependencies various ways, Planemo is configured with opinionated defaults geared at making building CWL tools that target Conda as easy as possible and build tools with requirements compatible with cwltool and Toil when running outside containers.
During the tool development introductory tutorial, we called planemo tool_init
with the argument --requirement seqtk@1.2 and the resulting tool contained such
a SoftwareRequirement in the form the following the YAML fragment:
SoftwareRequirement:
packages:
- package: seqtk
version:
- "1.2"
Planemo (and cwltool and Toil) can interpret these SoftwareRequirement annotations in various ways
including as Conda packages. When interpreting these as Conda packages
these runtimes can setup isolated, reproducible Conda environments for tool execution with the correct
packages installed (e.g. seqtk in the above example).
Note
Why Conda?
Many different package managers could potentially be targeted here, but we focus on Conda for a few key reasons.
No compilation at install time - binaries with their dependencies and libraries
Support for all operating systems
Easy to manage multiple versions of the same recipe
HPC-ready: no root privileges needed
Easy-to-write YAML recipes
Viberant communities
Note
Conda Terminology
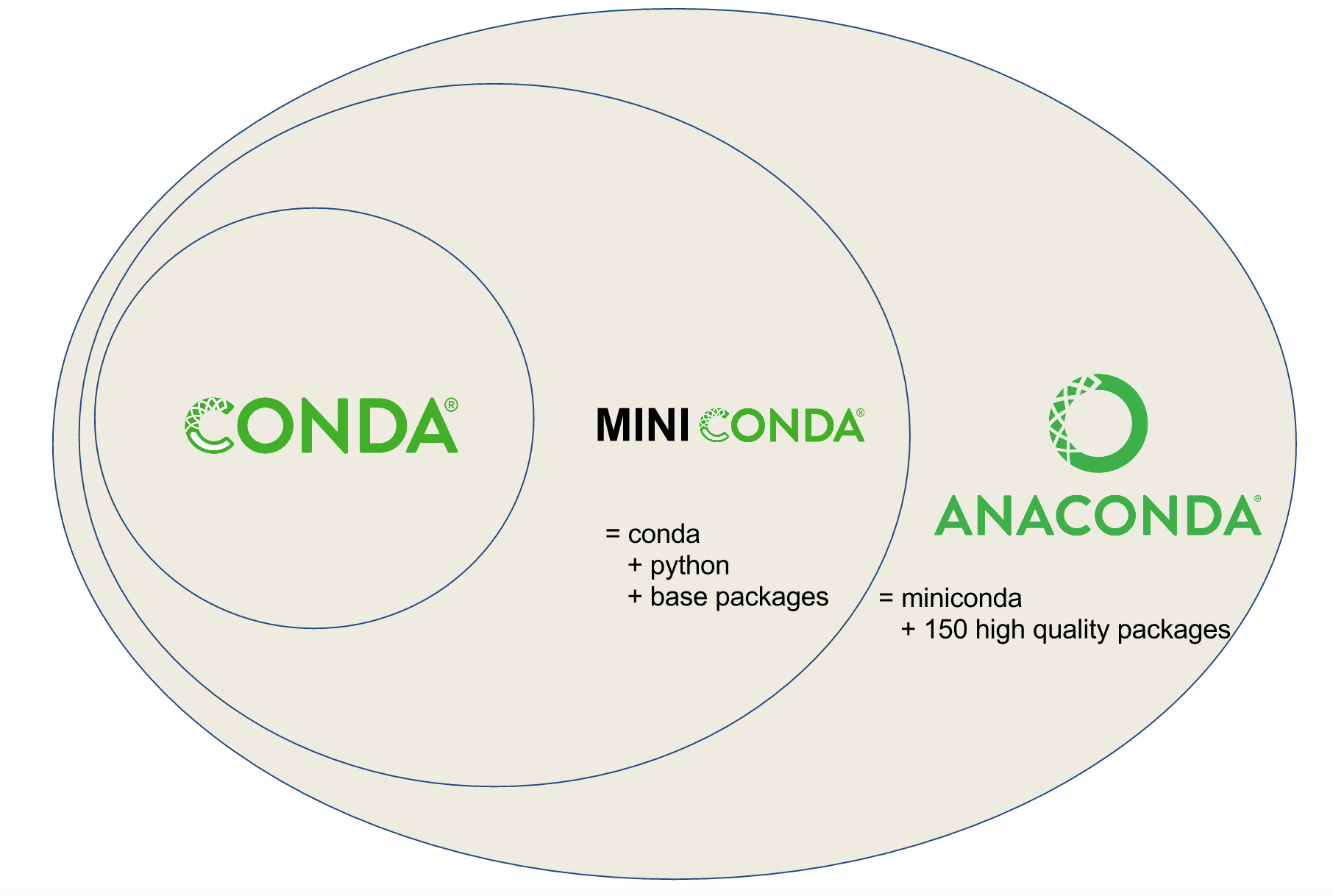
Conda recipes build packages that are published to channels.
Planemo is setup to target a few channels by default, these include iuc, bioconda,
conda_forge, defaults - the whole dependency management scheme outlined here works a lot
better if packages can be found in one of these “best practice” channels.
We can check if the requirements on a tool are available in best practice
Conda channels using an extended form of the planemo lint command (planemo lint was
introduced in the introductory tutorial). Passing --conda_requirements flag will ensure all
listed requirements are found.
$ planemo lint --conda_requirements seqtk_seq.cwl
Linting tool /Users/john/workspace/planemo/docs/writing/seqtk_seq.cwl
...
Applying linter requirements_in_conda... CHECK
.. INFO: Requirement [seqtk@1.2] matches target in best practice Conda channel [https://conda.anaconda.org/bioconda/osx-64].
Note
You can download a more complete version of the CWL seqtk seq from the Planemo tutorial using the command:
$ planemo project_init --template=seqtk_complete_cwl seqtk_example
$ cd seqtk_example
We can verify these tool requirements install with the conda_install command. With
its default parameters conda_install processes tools and creates isolated environments
for their declared Software Requirements (mirroring what can be done in production with
cwltool and Toil).
$ planemo conda_install seqtk_seq.cwl
Install conda target CondaTarget[seqtk,version=1.2]
/home/john/miniconda3/bin/conda create -y --name __seqtk@1.2 seqtk=1.2
Fetching package metadata ...............
Solving package specifications: ..........
Package plan for installation in environment /home/john/miniconda2/envs/__seqtk@1.2:
The following packages will be downloaded:
package | build
---------------------------|-----------------
seqtk-1.2 | 0 29 KB bioconda
The following NEW packages will be INSTALLED:
seqtk: 1.2-0 bioconda
zlib: 1.2.8-3
Fetching packages ...
seqtk-1.2-0.ta 100% |#############################################################| Time: 0:00:00 444.71 kB/s
Extracting packages ...
[ COMPLETE ]|################################################################################| 100%
Linking packages ...
[ COMPLETE ]|################################################################################| 100%
#
# To activate this environment, use:
# > source activate __seqtk@1.2
#
# To deactivate this environment, use:
# > source deactivate __seqtk@1.2
#
$ which seqtk
seqtk not found
$
The above install worked properly, but seqtk is not on your PATH because this merely
created an environment within the Conda directory for the seqtk installation. Planemo
will configure cwltool during testing to reuse this environment. If you wish to interactively explore
the resulting enviornment to explore the installed tool or produce test data the output
of the conda_env command can be sourced.
$ . <(planemo conda_env seqtk_seq.cwl)
Deactivate environment with conda_env_deactivate
(seqtk_seq) $ which seqtk
/home/planemo/miniconda3/envs/jobdepsiJClEUfecc6d406196737781ff4456ec60975c137e04884e4f4b05dc68192f7cec4656/bin/seqtk
(seqtk_seq) $ seqtk seq
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
-U convert all bases to uppercases
-S strip of white spaces in sequences
(seqtk_seq) $ conda_env_deactivate
$
As shown above the conda_env_deactivate will be created in this environment and can
be used to restore your initial shell configuration.
Here is a portion of the output from the testing command planemo test seqtk_seq.cwl
demonstrating using this tool.
$ planemo test --no-container seqtk_seq.cwl
Enable beta testing mode for testing.
cwltool INFO: /Users/john/workspace/planemo/.venv/bin/planemo 1.0.20170828135420
cwltool INFO: Resolved '/Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl' to 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl'
cwltool INFO: [job seqtk_seq.cwl] /private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpaDQ1nK$ seqtk \
seq \
-a \
/private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpJtPKCr/stg24cf7e67-5ca6-44a4-a46b-26cbe104e1d4/2.fastq > /private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpaDQ1nK/out
cwltool INFO: [job seqtk_seq.cwl] completed success
cwltool INFO: Final process status is success
galaxy.tools.parser.factory INFO: Loading CWL tool - this is experimental - tool likely will not function in future at least in same way.
All 1 test(s) executed passed.
seqtk_seq_0: passed
Since seqtk isn’t on the path and we did not use a container, we can see the SoftwareRequirement
resolution was successful and it found the environment we previously installed with conda_install.
This can be used outside of Planemo testing as well, the following invocation shows running a job with cwltool using an environment like the one created above:
$ cwltool --no-container --beta-conda-dependencies seqtk_seq.cwl seqtk_seq_job.yml
/Users/john/workspace/planemo/.venv/bin/cwltool 1.0.20180508202931
Resolved 'seqtk_seq.cwl' to 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl'
No handlers could be found for logger "rdflib.term"
[job seqtk_seq.cwl] /private/tmp/docker_tmpDQYeqC$ seqtk \
seq \
-a \
/private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpQwBqPo/stg8cf2282a-d807-4f90-b94d-feeda004cacd/2.fastq > /private/tmp/docker_tmpDQYeqC/out
PREFIX=/Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/cwltool_deps/_conda
installing: python-3.6.3-h47c878a_7 ...
Python 3.6.3 :: Anaconda, Inc.
installing: ca-certificates-2017.08.26-ha1e5d58_0 ...
installing: conda-env-2.6.0-h36134e3_0 ...
installing: libcxxabi-4.0.1-hebd6815_0 ...
installing: tk-8.6.7-h35a86e2_3 ...
installing: xz-5.2.3-h0278029_2 ...
installing: yaml-0.1.7-hc338f04_2 ...
installing: zlib-1.2.11-hf3cbc9b_2 ...
installing: libcxx-4.0.1-h579ed51_0 ...
installing: openssl-1.0.2n-hdbc3d79_0 ...
installing: libffi-3.2.1-h475c297_4 ...
installing: ncurses-6.0-hd04f020_2 ...
installing: libedit-3.1-hb4e282d_0 ...
installing: readline-7.0-hc1231fa_4 ...
installing: sqlite-3.20.1-h7e4c145_2 ...
installing: asn1crypto-0.23.0-py36h782d450_0 ...
installing: certifi-2017.11.5-py36ha569be9_0 ...
installing: chardet-3.0.4-py36h96c241c_1 ...
installing: idna-2.6-py36h8628d0a_1 ...
installing: pycosat-0.6.3-py36hee92d8f_0 ...
installing: pycparser-2.18-py36h724b2fc_1 ...
installing: pysocks-1.6.7-py36hfa33cec_1 ...
installing: python.app-2-py36h54569d5_7 ...
installing: ruamel_yaml-0.11.14-py36h9d7ade0_2 ...
installing: six-1.11.0-py36h0e22d5e_1 ...
installing: cffi-1.11.2-py36hd3e6348_0 ...
installing: setuptools-36.5.0-py36h2134326_0 ...
installing: cryptography-2.1.4-py36h842514c_0 ...
installing: wheel-0.30.0-py36h5eb2c71_1 ...
installing: pip-9.0.1-py36h1555ced_4 ...
installing: pyopenssl-17.5.0-py36h51e4350_0 ...
installing: urllib3-1.22-py36h68b9469_0 ...
installing: requests-2.18.4-py36h4516966_1 ...
installing: conda-4.3.31-py36_0 ...
installation finished.
Fetching package metadata .................
Solving package specifications: .
Package plan for installation in environment /Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/cwltool_deps/_conda:
The following packages will be UPDATED:
conda: 4.3.31-py36_0 --> 4.3.33-py36_0 conda-forge
conda-4.3.33-p 100% |#################################################################| Time: 0:00:00 1.13 MB/s
Package plan for installation in environment /Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/cwltool_deps/_conda/envs/__seqtk@1.2:
The following NEW packages will be INSTALLED:
seqtk: 1.2-1 bioconda
zlib: 1.2.11-0 conda-forge
[job seqtk_seq.cwl] completed success
{
"output1": {
"checksum": "sha1$322e001e5a99f19abdce9f02ad0f02a17b5066c2",
"basename": "out",
"location": "file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/out",
"path": "/Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/out",
"class": "File",
"size": 150
}
}
Final process status is success
This demonstrates that cwltool will install the packages needed on the first run, if we rerun cwltool it will reuse that previous environment.
$ cwltool --no-container --beta-conda-dependencies seqtk_seq.cwl seqtk_seq_job.yml
/Users/john/workspace/planemo/.venv/bin/cwltool 1.0.20180508202931
Resolved 'seqtk_seq.cwl' to 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl'
No handlers could be found for logger "rdflib.term"
[job seqtk_seq.cwl] /private/tmp/docker_tmp4vvE_i$ seqtk \
seq \
-a \
/private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpcvQ3Ph/stg2ef3a21c-9fb0-4099-88c2-36e24719901d/2.fastq > /private/tmp/docker_tmp4vvE_i/out
[job seqtk_seq.cwl] completed success
{
"output1": {
"checksum": "sha1$322e001e5a99f19abdce9f02ad0f02a17b5066c2",
"basename": "out",
"location": "file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/out",
"path": "/Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/out",
"class": "File",
"size": 150
}
}
Final process status is success
And the same thing is possible with Toil.
$ cwltoil --no-container --beta-conda-dependencies seqtk_seq.cwl seqtk_seq_job.yml
jlaptop17.local 2018-05-23 15:27:25,754 MainThread INFO toil.lib.bioio: Root logger is at level 'INFO', 'toil' logger at level 'INFO'.
jlaptop17.local 2018-05-23 15:27:25,785 MainThread INFO toil.jobStores.abstractJobStore: The workflow ID is: '92328fb2-33b7-44cd-879f-41d8cbf94555'
Resolved 'seqtk_seq.cwl' to 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl'
jlaptop17.local 2018-05-23 15:27:25,787 MainThread INFO cwltool: Resolved 'seqtk_seq.cwl' to 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl'
jlaptop17.local 2018-05-23 15:27:27,002 MainThread WARNING rdflib.term: http://schema.org/docs/!DOCTYPE html does not look like a valid URI, trying to serialize this will break.
jlaptop17.local 2018-05-23 15:27:27,396 MainThread INFO rdflib.plugins.parsers.pyRdfa: Current options:
preserve space : True
output processor graph : True
output default graph : True
host language : RDFa Core
accept embedded RDF : False
check rdfa lite : False
cache vocabulary graphs : False
jlaptop17.local 2018-05-23 15:27:29,797 MainThread INFO toil.common: Using the single machine batch system
jlaptop17.local 2018-05-23 15:27:29,798 MainThread WARNING toil.batchSystems.singleMachine: Limiting maxCores to CPU count of system (8).
jlaptop17.local 2018-05-23 15:27:29,798 MainThread WARNING toil.batchSystems.singleMachine: Limiting maxMemory to physically available memory (17179869184).
jlaptop17.local 2018-05-23 15:27:29,808 MainThread INFO toil.common: Created the workflow directory at /var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/toil-92328fb2-33b7-44cd-879f-41d8cbf94555-132281828025877
jlaptop17.local 2018-05-23 15:27:29,808 MainThread WARNING toil.batchSystems.singleMachine: Limiting maxDisk to physically available disk (202669449216).
jlaptop17.local 2018-05-23 15:27:29,815 MainThread INFO toil.common: User script ModuleDescriptor(dirPath='/Users/john/workspace/planemo/.venv/lib/python2.7/site-packages', name='toil.cwl.cwltoil', fromVirtualEnv=True) belongs to Toil. No need to auto-deploy it.
jlaptop17.local 2018-05-23 15:27:29,816 MainThread INFO toil.common: No user script to auto-deploy.
jlaptop17.local 2018-05-23 15:27:29,816 MainThread INFO toil.common: Written the environment for the jobs to the environment file
jlaptop17.local 2018-05-23 15:27:29,816 MainThread INFO toil.common: Caching all jobs in job store
jlaptop17.local 2018-05-23 15:27:29,816 MainThread INFO toil.common: 0 jobs downloaded.
jlaptop17.local 2018-05-23 15:27:29,911 MainThread INFO toil: Running Toil version 3.15.0-0e3a87e738f5e0e7cff64bfdad337d592bd92704.
jlaptop17.local 2018-05-23 15:27:29,911 MainThread INFO toil.realtimeLogger: Real-time logging disabled
jlaptop17.local 2018-05-23 15:27:29,937 MainThread INFO toil.toilState: (Re)building internal scheduler state
2018-05-23 15:27:29,937 - toil.toilState - INFO - (Re)building internal scheduler state
jlaptop17.local 2018-05-23 15:27:29,938 MainThread INFO toil.leader: Found 1 jobs to start and 0 jobs with successors to run
2018-05-23 15:27:29,938 - toil.leader - INFO - Found 1 jobs to start and 0 jobs with successors to run
jlaptop17.local 2018-05-23 15:27:29,938 MainThread INFO toil.leader: Checked batch system has no running jobs and no updated jobs
2018-05-23 15:27:29,938 - toil.leader - INFO - Checked batch system has no running jobs and no updated jobs
jlaptop17.local 2018-05-23 15:27:29,938 MainThread INFO toil.leader: Starting the main loop
2018-05-23 15:27:29,938 - toil.leader - INFO - Starting the main loop
jlaptop17.local 2018-05-23 15:27:29,939 MainThread INFO toil.leader: Issued job 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl' seqtk seq e/V/jobsxUpTU with job batch system ID: 0 and cores: 1, disk: 3.0 G, and memory: 2.0 G
2018-05-23 15:27:29,939 - toil.leader - INFO - Issued job 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl' seqtk seq e/V/jobsxUpTU with job batch system ID: 0 and cores: 1, disk: 3.0 G, and memory: 2.0 G
jlaptop17.local 2018-05-23 15:27:31,409 MainThread INFO toil.leader: Job ended successfully: 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl' seqtk seq e/V/jobsxUpTU
2018-05-23 15:27:31,409 - toil.leader - INFO - Job ended successfully: 'file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/seqtk_seq.cwl' seqtk seq e/V/jobsxUpTU
jlaptop17.local 2018-05-23 15:27:31,411 MainThread INFO toil.leader: Finished the main loop: no jobs left to run
2018-05-23 15:27:31,411 - toil.leader - INFO - Finished the main loop: no jobs left to run
jlaptop17.local 2018-05-23 15:27:31,411 MainThread INFO toil.serviceManager: Waiting for service manager thread to finish ...
2018-05-23 15:27:31,411 - toil.serviceManager - INFO - Waiting for service manager thread to finish ...
jlaptop17.local 2018-05-23 15:27:31,946 MainThread INFO toil.serviceManager: ... finished shutting down the service manager. Took 0.535056114197 seconds
2018-05-23 15:27:31,946 - toil.serviceManager - INFO - ... finished shutting down the service manager. Took 0.535056114197 seconds
jlaptop17.local 2018-05-23 15:27:31,947 MainThread INFO toil.statsAndLogging: Waiting for stats and logging collator thread to finish ...
2018-05-23 15:27:31,947 - toil.statsAndLogging - INFO - Waiting for stats and logging collator thread to finish ...
jlaptop17.local 2018-05-23 15:27:31,960 MainThread INFO toil.statsAndLogging: ... finished collating stats and logs. Took 0.0131621360779 seconds
2018-05-23 15:27:31,960 - toil.statsAndLogging - INFO - ... finished collating stats and logs. Took 0.0131621360779 seconds
jlaptop17.local 2018-05-23 15:27:31,961 MainThread INFO toil.leader: Finished toil run successfully
2018-05-23 15:27:31,961 - toil.leader - INFO - Finished toil run successfully
{
"output1": {
"checksum": "sha1$322e001e5a99f19abdce9f02ad0f02a17b5066c2",
"basename": "out",
"nameext": "",
"nameroot": "out",
"http://commonwl.org/cwltool#generation": 0,
"location": "file:///Users/john/workspace/planemo/project_templates/seqtk_complete_cwl/out",
"class": "File",
"size": 150
}
jlaptop17.local 2018-05-23 15:27:31,972 MainThread INFO toil.common: Successfully deleted the job store: <toil.jobStores.fileJobStore.FileJobStore object at 0x10554d490>
}2018-05-23 15:27:31,972 - toil.common - INFO - Successfully deleted the job store: <toil.jobStores.fileJobStore.FileJobStore object at 0x10554d490>
Finding Existing Conda Packages
How did we know what software name and software version to use? We found the existing
packages available for Conda and referenced them. To do this yourself, you can simply
use the planemo command conda_search. If we do a search for seqt it will show
all the software and all the versions available matching that search term - including
seqtk.
$ planemo conda_search seqt
/Users/john/miniconda3/bin/conda search --override-channels --channel iuc --channel conda-forge --channel bioconda --channel defaults '*seqt*'
Loading channels: done
# Name Version Build Channel
bioconductor-htseqtools 1.26.0 r3.4.1_0 bioconda
bioconductor-seqtools 1.10.0 r3.3.2_0 bioconda
bioconductor-seqtools 1.10.0 r3.4.1_0 bioconda
bioconductor-seqtools 1.12.0 r3.4.1_0 bioconda
seqtk r75 0 bioconda
seqtk r82 0 bioconda
seqtk r82 1 bioconda
seqtk r93 0 bioconda
seqtk 1.2 0 bioconda
seqtk 1.2 1 bioconda
Note
The Planemo command conda_search is a light wrapper around the underlying
conda search command but configured to use the same channels and other options as
Planemo and Galaxy. The following Conda command would also work to search:
$ $HOME/miniconda3/bin/conda -c iuc -c conda-forge -c bioconda '*seqt*'
For Conda versions 4.3.X or less, the search invocation would be something a bit different:
$ $HOME/miniconda3/bin/conda -c iuc -c conda-forge -c bioconda seqt
Alternatively the Anaconda website can be used to search for packages. Typing seqtk
into the search form on that page and clicking the top result will bring on to this page with information about the Bioconda package.
When using the website to search though, you need to aware of what channel you are using. By default, Planemo and Galaxy will search a few different Conda channels. While it is possible to configure a local Planemo or Galaxy to target different channels - the current best practice is to add tools to the existing channels.
The existing channels include:
Exercise - Leveraging Bioconda
Use the project_init command to download this exercise.
$ planemo project_init --template conda_exercises_cwl conda_exercises
$ cd conda_exercises/exercise_1
$ ls
pear.cwl test-data
This project template contains a few exercises. The first uses a CWL tool for
PEAR - Paired-End reAd mergeR.
This tool however has no SoftwareRequirement or container annotations and so will
not work properly without modification.
Run
planemo test pear.cwlto verify the tool does not function without dependencies defined.Use
--conda_requirementsflag withplanemo lintto verify it does indeed lack requirements.Use
planemo conda_searchor the Anaconda website to search for the correct package and version in a best practice channel.Update
pear.cwlwith the correctSoftwareRequirementhints.Re-run the
lintcommand from above to verify the tool now has the correct dependency definition.Re-run the
testcommand from above to verify the tool test now works properly.
Building New Conda Packages
Frequently packages your tool will require are not found in Bioconda or conda-forge yet. In these cases, it is likely best to contribute your package to one of these projects. Unless the tool is exceedingly general Bioconda is usually the correct starting point.
Note
Many things that are not strictly or even remotely “bio” have been accepted into Bioconda - including tools for image analysis, natural language processing, and cheminformatics.
To get quickly learn to write Conda recipes for typical Galaxy tools, please read the following pieces of external documentation.
Contributing to Bioconda in particular focusing on
Contributing a recipe (through “Write a Recipe”)
Building conda packages in particular
Building conda packages with conda skeleton (the best approach for common scripting languages such as R and Python)
Then return to the Bioconda documentation and read
The rest of “Contributing a recipe” continuing from Testing locally
And finally Guidelines for bioconda recipes
These guidelines in particular can be skimmed depending on your recipe type, for instance that document provides specific advice for:
To go a little deeper, you may want to read:
And finally to debug problems the Bioconda troubleshooting documentation may prove useful.
Exercise - Build a Recipe
If you have just completed the exercise above - this exercise can be found in parent folder. Get
there with cd ../exercise_2. If not, the exercise can be downloaded with
$ planemo project_init --template conda_exercises_cwl conda_exercises
$ cd conda_exercises/exercise_2
$ ls
fleeqtk_seq.cwl fleeqtk_seq_tests.yml test-data
This is the skeleton of a tool wrapping the parody bioinformatics software package fleeqtk.
fleeqtk is a fork of the project seqtk that many Planemo tutorials are built around and the
example tool should hopefully be fairly familiar. fleeqtk version 1.3 can be downloaded
from here and built using
make. The result of make includes a single executable fleeqtk.
Clone and branch Bioconda.
Build a recipe for fleeqtk version 1.3. You may wish to start from scratch (
conda skeletonis not available for C programs like fleeqtk), or copy the recipe of seqtk and modify it for fleeqtk.Use
conda buildor Bioconda tooling to build the recipe.Run
planemo test --conda_use_local fleeqtk_seq.cwlto verify the resulting package works as expected.
Congratulations on writing a Conda recipe and building a package! Upon succesfully building and testing such a Bioconda package, you would normally push your branch to Github and open a pull request. This step is skipped here as to not pollute Bioconda with unneeded software packages.
Dependencies and Containers
Note
This section is a continuation of Dependencies and Conda, please review that section for background information on resolving Software Requirements with Conda.
Common Workflow Language tools can be annotated with arbitrary Docker requirements, see the CWL User Guide for a discussion about how to do this in general.
This document will discuss some techniques to find containers automatically from
the SoftwareRequirement annotations when using Planemo, cwltool, or Toil.
You will ultimately want to explicitly annotate your tools with the containers
we describe here so that other CWL implementations will be able to find containers
for your tool, but there are real advantages to using these containers instead
of ad-hoc things you may build with a Dockerfile.
They provide superior reproducibility because the same binary Conda packages will automatically be used for both bare metal dependencies and inside containers.
They are constructed automatically from existing Conda packages so you as a tool developer won’t need to write
Dockerfiles or register projects on Docker Hub.They are produced using mulled which produce very small containers that make deployment easier regardless of the CWL implementation you are using.
Annotating Software Requirements reduces the opaqueness of the Docker process. With this method it is entirely traceable how the container was constructed from what sources were fetched, which exact build of every dependency was used, to how packages in the container were built. Beyond that metadata about the packages can be fetched from Bioconda (e.g. this).
Read more about this reproducibility stack in our preprint Practical computational reproducibility in the life sciences.
BioContainers
Note
This section is a continuation of Dependencies and Conda, please review that section for background information on resolving Software Requirements with Conda.
Finding BioContainers
If a tool contains Software Requirements in best practice Conda channels, a BioContainers-style container can be found or built for it.
As reminder, planemo lint --conda_requirements <tool.cwl> can be used
to check if a tool contains only best-practice requirement tags. The lint
command can also be fed the --biocontainers flag to check if a
BioContainers container has been registered that is compatible with that tool.
This last linter indicates that indeed a container has been registered
that is compatible with this tool – quay.io/biocontainers/seqtk:1.2--1.
We didn’t do any extra work to build this container for this tool, all
Bioconda recipes are packaged into containers and registered on quay.io
as part of the BioContainers project.
This tool can be tested using planemo test in its BioContainer
Docker container using the flag --biocontainers as shown below.
The Conda exercises project template has an example tool (exercise3) that we
can use to demonstrate --biocontainers. If you are continuing from the Conda
tutorial, simply move to ../exercise3 otherwise using planemo project_init
to grab the exercise as show below.
$ planemo project_init --template conda_exercises_cwl conda_exercises
$ cd conda_exercises/exercise3
$ planemo lint --biocontainers seqtk_seq.cwl
Linting tool /home/planemo/conda_exercises_cwl/exercise_3/seqtk_seq.cwl
Applying linter general... CHECK
.. CHECK: Tool defines a version [0.0.1].
.. CHECK: Tool defines a name [Convert to FASTA (seqtk)].
.. CHECK: Tool defines an id [seqtk_seq].
.. CHECK: Tool specifies profile version [16.04].
Applying linter cwl_validation... CHECK
.. INFO: CWL appears to be valid.
Applying linter docker_image... WARNING
.. WARNING: Tool does not specify a DockerPull source.
Applying linter new_draft... CHECK
.. INFO: Modern CWL version [v1.0]
Applying linter biocontainer_registered... CHECK
.. INFO: BioContainer best-practice container found [quay.io/biocontainers/seqtk:1.2--1].
Failed linting
$ planemo test --biocontainers seqtk_seq.cwl
Enable beta testing mode for testing.
cwltool INFO: /Users/john/workspace/planemo/.venv/bin/planemo 1.0.20180508202931
cwltool INFO: Resolved '/Users/john/workspace/planemo/project_templates/conda_exercises_cwl/exercise_3/seqtk_seq.cwl' to 'file:///Users/john/workspace/planemo/project_templates/conda_exercises_cwl/exercise_3/seqtk_seq.cwl'
galaxy.tools.deps.containers INFO: Checking with container resolver [ExplicitContainerResolver[]] found description [None]
galaxy.tools.deps.containers INFO: Checking with container resolver [CachedMulledDockerContainerResolver[namespace=biocontainers]] found description [None]
galaxy.tools.deps.containers INFO: Checking with container resolver [MulledDockerContainerResolver[namespace=biocontainers]] found description [ContainerDescription[identifier=quay.io/biocontainers/seqtk:1.2--1,type=docker]]
cwltool INFO: [job seqtk_seq.cwl] /private/tmp/docker_tmpMEipaU$ docker \
run \
-i \
--volume=/private/tmp/docker_tmpMEipaU:/private/tmp/docker_tmpMEipaU:rw \
--volume=/private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpxkm9dp:/tmp:rw \
--volume=/Users/john/workspace/planemo/project_templates/conda_exercises_cwl/exercise_3/test-data/2.fastq:/private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpjAVM_1/stgddf6fc2a-dd13-4322-9b88-68571a1697dd/2.fastq:ro \
--workdir=/private/tmp/docker_tmpMEipaU \
--read-only=true \
--log-driver=none \
--user=502:20 \
--rm \
--env=TMPDIR=/tmp \
--env=HOME=/private/tmp/docker_tmpMEipaU \
quay.io/biocontainers/seqtk:1.2--1 \
seqtk \
seq \
-a \
/private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpjAVM_1/stgddf6fc2a-dd13-4322-9b88-68571a1697dd/2.fastq > /private/tmp/docker_tmpMEipaU/out
cwltool INFO: [job seqtk_seq.cwl] completed success
cwltool INFO: Final process status is success
All 1 test(s) executed passed.
seqtk_seq_0: passed
Exercise - Leveraging Bioconda
Try the above command without the
--biocontainersargument. Verify the tool does not run in a container by default.Add a DockerRequirement based on the the lint output above to annotate this tool with a Biocontainers Docker container and rerun test to verify the tool works now.
Building BioContainers
In this seqtk example above the relevant BioContainer already existed on quay.io, this won’t always be the case. For tools that contain multiple Software Requirements tags an existing container likely won’t exist. The mulled toolkit (distributed with planemo or available standalone) can be used to build containers for such tools. For such tools, if cwltool or Toil is configured to use BioContainers it will attempt to build these containers on the fly by default (though this behavior can be disabled).
You can try it directly using the mull command in Planemo. The conda_testing
Planemo project template has a toy example tool with two requirements for
demonstrating this - bwa_and_samtools.cwl.
$ planemo project_init --template=conda_testing_cwl conda_testing
$ cd conda_testing/
$ planemo mull bwa_and_samtools.cwl
/Users/john/.planemo/involucro -v=3 -f /Users/john/workspace/planemo/.venv/lib/python2.7/site-packages/galaxy_lib-17.9.0-py2.7.egg/galaxy/tools/deps/mulled/invfile.lua -set CHANNELS='iuc,bioconda,r,defaults,conda-forge' -set TEST='true' -set TARGETS='samtools=1.3.1,bwa=0.7.15' -set REPO='quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820' -set BINDS='build/dist:/usr/local/' -set PREINSTALL='conda install --quiet --yes conda=4.3' build
/Users/john/.planemo/involucro -v=3 -f /Users/john/workspace/planemo/.venv/lib/python2.7/site-packages/galaxy_lib-17.9.0-py2.7.egg/galaxy/tools/deps/mulled/invfile.lua -set CHANNELS='iuc,bioconda,r,defaults,conda-forge' -set TEST='true' -set TARGETS='samtools=1.3.1,bwa=0.7.15' -set REPO='quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820' -set BINDS='build/dist:/usr/local/' -set PREINSTALL='conda install --quiet --yes conda=4.3' build
[Jun 19 11:28:35] DEBU Run file [/Users/john/workspace/planemo/.venv/lib/python2.7/site-packages/galaxy_lib-17.9.0-py2.7.egg/galaxy/tools/deps/mulled/invfile.lua]
[Jun 19 11:28:35] STEP Run image [continuumio/miniconda:latest] with command [[rm -rf /data/dist]]
[Jun 19 11:28:35] DEBU Creating container [step-730a02d79e]
[Jun 19 11:28:35] DEBU Created container [5e4b5f83c455 step-730a02d79e], starting it
[Jun 19 11:28:35] DEBU Container [5e4b5f83c455 step-730a02d79e] started, waiting for completion
[Jun 19 11:28:36] DEBU Container [5e4b5f83c455 step-730a02d79e] completed with exit code [0] as expected
[Jun 19 11:28:36] DEBU Container [5e4b5f83c455 step-730a02d79e] removed
[Jun 19 11:28:36] STEP Run image [continuumio/miniconda:latest] with command [[/bin/sh -c conda install --quiet --yes conda=4.3 && conda install -c iuc -c bioconda -c r -c defaults -c conda-forge samtools=1.3.1 bwa=0.7.15 -p /usr/local --copy --yes --quiet]]
[Jun 19 11:28:36] DEBU Creating container [step-e95bf001c8]
[Jun 19 11:28:36] DEBU Created container [72b9ca0e56f8 step-e95bf001c8], starting it
[Jun 19 11:28:37] DEBU Container [72b9ca0e56f8 step-e95bf001c8] started, waiting for completion
[Jun 19 11:28:46] SOUT Fetching package metadata .........
[Jun 19 11:28:47] SOUT Solving package specifications: .
[Jun 19 11:28:50] SOUT
[Jun 19 11:28:50] SOUT Package plan for installation in environment /opt/conda:
[Jun 19 11:28:50] SOUT
[Jun 19 11:28:50] SOUT The following packages will be UPDATED:
[Jun 19 11:28:50] SOUT
[Jun 19 11:28:50] SOUT conda: 4.3.11-py27_0 --> 4.3.22-py27_0
[Jun 19 11:28:50] SOUT
[Jun 19 11:29:04] SOUT Fetching package metadata .................
[Jun 19 11:29:06] SOUT Solving package specifications: .
[Jun 19 11:29:56] SOUT
[Jun 19 11:29:56] SOUT Package plan for installation in environment /usr/local:
[Jun 19 11:29:56] SOUT
[Jun 19 11:29:56] SOUT The following NEW packages will be INSTALLED:
[Jun 19 11:29:56] SOUT
[Jun 19 11:29:56] SOUT bwa: 0.7.15-1 bioconda
[Jun 19 11:29:56] SOUT curl: 7.52.1-0
[Jun 19 11:29:56] SOUT libgcc: 5.2.0-0
[Jun 19 11:29:56] SOUT openssl: 1.0.2l-0
[Jun 19 11:29:56] SOUT pip: 9.0.1-py27_1
[Jun 19 11:29:56] SOUT python: 2.7.13-0
[Jun 19 11:29:56] SOUT readline: 6.2-2
[Jun 19 11:29:56] SOUT samtools: 1.3.1-5 bioconda
[Jun 19 11:29:56] SOUT setuptools: 27.2.0-py27_0
[Jun 19 11:29:56] SOUT sqlite: 3.13.0-0
[Jun 19 11:29:56] SOUT tk: 8.5.18-0
[Jun 19 11:29:56] SOUT wheel: 0.29.0-py27_0
[Jun 19 11:29:56] SOUT zlib: 1.2.8-3
[Jun 19 11:29:56] SOUT
[Jun 19 11:29:57] DEBU Container [72b9ca0e56f8 step-e95bf001c8] completed with exit code [0] as expected
[Jun 19 11:29:57] DEBU Container [72b9ca0e56f8 step-e95bf001c8] removed
[Jun 19 11:29:57] STEP Wrap [build/dist] as [quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0]
[Jun 19 11:29:57] DEBU Creating container [step-6f1c176372]
[Jun 19 11:29:58] DEBU Packing succeeded
As the output indicates, this command built the container named
quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0.
This is the same namespace / URL that would be used if or when published by
the BioContainers project.
Note
The first part of this mulled-v2 hash is a hash of the package names
that went into it, the second the packages used and build number. Check out
the Multi-package Containers
web application to explore best practice channels and build such hashes.
We can see this new container when running the Docker command images and
explore the new container interactively with docker run.
$ docker images
REPOSITORY TAG IMAGE ID CREATED SIZE
quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40 03dc1d2818d9de56938078b8b78b82d967c1f820-0 a740fe1e6a9e 16 hours ago 104 MB
quay.io/biocontainers/seqtk 1.2--0 10bc359ebd30 2 days ago 7.34 MB
continuumio/miniconda latest 6965a4889098 3 weeks ago 437 MB
bgruening/busybox-bash 0.1 3d974f51245c 9 months ago 6.73 MB
$ docker run -i -t quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0 /bin/bash
bash-4.2# which samtools
/usr/local/bin/samtools
bash-4.2# which bwa
/usr/local/bin/bwa
As before, we can test running the tool inside its container in cwltool using
the --biocontainers flag.
$ planemo test --biocontainers bwa_and_samtools.cwl
Enable beta testing mode for testing.
cwltool INFO: /Users/john/workspace/planemo/.venv/bin/planemo 1.0.20180508202931
cwltool INFO: Resolved '/Users/john/workspace/planemo/project_templates/conda_testing_cwl/bwa_and_samtools.cwl' to 'file:///Users/john/workspace/planemo/project_templates/conda_testing_cwl/bwa_and_samtools.cwl'
galaxy.tools.deps.containers INFO: Checking with container resolver [ExplicitContainerResolver[]] found description [None]
galaxy.tools.deps.containers INFO: Checking with container resolver [CachedMulledDockerContainerResolver[namespace=biocontainers]] found description [ContainerDescription[identifier=quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0,type=docker]]
cwltool INFO: [job bwa_and_samtools.cwl] /private/tmp/docker_tmpYJnmO4$ docker \
run \
-i \
--volume=/private/tmp/docker_tmpYJnmO4:/private/tmp/docker_tmpYJnmO4:rw \
--volume=/private/var/folders/78/zxz5mz4d0jn53xf0l06j7ppc0000gp/T/tmpVI06me:/tmp:rw \
--workdir=/private/tmp/docker_tmpYJnmO4 \
--read-only=true \
--user=502:20 \
--rm \
--env=TMPDIR=/tmp \
--env=HOME=/private/tmp/docker_tmpYJnmO4 \
quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0 \
sh \
-c \
'bwa > bwa_help.txt 2>&1; samtools > samtools_help.txt 2>&1'
cwltool INFO: [job bwa_and_samtools.cwl] completed success
cwltool INFO: Final process status is success
All 1 test(s) executed passed.
bwa_and_samtools_0: passed
In particular take note of the line:
2017-03-01 10:20:59,142 INFO [galaxy.tools.deps.containers] Checking with container resolver [CachedMulledDockerContainerResolver[namespace=biocontainers]] found description [ContainerDescription[identifier=quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0,type=docker]]
Here we can see the container ID (quay.io/biocontainers/mulled-v2-fe8faa35dbf6dc65a0f7f5d4ea12e31a79f73e40:03dc1d2818d9de56938078b8b78b82d967c1f820-0)
from earlier has been cached on our Docker host is picked up by cwltool. This is used to run the
simple tool tests and indeed they pass.
In our initial seqtk example, the container resolver that matched was of type
MulledDockerContainerResolver indicating that the Docker image would be downloaded
from the BioContainers repository and this time the resolve that matched was of type
CachedMulledDockerContainerResolver meaning that cwltool would just use the locally
cached version from the Docker host (i.e. the one we built with planemo mull
above).
Note
Planemo doesn’t yet expose options that make it possible to build mulled
containers for local packages that have yet to be published to anaconda.org
but the mulled toolkit allows this. See mulled documentation for more
information. However, once a container for a local package is built with
mulled-build-tool the --biocontainers command should work to test
it.
Publishing BioContainers
Building unpublished BioContainers on the fly is great for testing but for production use and to increase reproducibility such containers should ideally be published as well.
BioContainers maintains a registry of package combinations to be published
using these long mulled hashes. This registry is represented as a Github repository
named multi-package-containers.
The Planemo command container_register will inspect a tool and open a
Github pull request to add the tool’s combination
of packages to the registry. Once merged, this pull request will
result in the corresponding BioContainers image to be published (with the
correct mulled has as its name) - these can be subsequently be picked up by
Galaxy.
Various Github related settings need to be configured in order for Planemo
to be able to open pull requests on your behalf as part of the
container_register command. To simplify all of this - the Planemo community
maintains a list of Github repositories containing Galaxy and/or CWL tools that
are scanned daily by Travis. For each such repository, the Travis job will run
container_register across the repository on all tools resulting in new registry
pull requests for all new combinations of tools. This list is maintained
in a script named monitor.sh in the planemo-monitor repository. The easiest way
to ensure new containers are built for your tools is simply to open open a pull
request to add your tool repositories to this list.